Tolerance and Autoimmunity
Introduction
The immune system protects against invasion by foreign organisms. However, immune responses can cause damage if they are directed against the individual’s own components, which are often referred to as self-antigens or autoantigens. Therefore, the immune system has developed mechanisms to allow it to respond to foreign antigens but not self-antigens. Both the innate and adaptive immune responses have evolved to make this distinction. The state of specific unresponsiveness to self-antigen is known as self-tolerance. This chapter focuses on the adaptive immune responses that are the basis for self-tolerance. The first part of this chapter will discuss the basic mechanisms of tolerance to self and how these mechanisms protect the host from harmful immune reactions. The second part of the chapter will discuss how mechanisms of tolerance can go awry, leading to the activation of autoreactive lymphocytes and the development of autoimmune disease.
Discovery of tolerance. A key finding by Ray Owen in 1945 was that tolerance to self-antigens was acquired. He studied cows that were dizygotic twins—they had different red blood types but shared a common blood supply in utero. He showed that, as adults, if one of the twins was injected with red blood cells from the other twin, it did not generate anti-red blood cell antibodies. Thus, it appeared that sharing of “foreign” blood cell antigens during the fetal stage had produced long-lasting tolerance.
These observations provided the basis for classic experiments in mice performed by Sir Peter Medawar and his colleagues in the 1950s, for which he was awarded a Nobel Prize in 1960. They observed that adult mice of one inbred strain (strain A) rejected skin grafts from mice of another strain (strain B); the critical difference between the two mouse strains was in the major histocompatibility complex (MHC) molecules they expressed. However, if mice of strain A were injected within 24 hours after birth with bone marrow stem cells from strain B, they did not reject skin grafts from the donor B strain when they grew to adulthood. These results suggested that when immature, developing lymphocytes are exposed to a foreign antigen, they fail to mount an immune response to the antigen. Instead, the antigen is accepted as self and the immune system becomes tolerant to it. This phenomenon became known as neonatal tolerance.
Self-tolerance, a property of both B and T lymphocytes, is a state of specific unresponsiveness to self-antigen. It occurs when the interaction of an autoantigen with self-antigen-specific lymphocytes fails to activate the lymphocytes. Thus, to be tolerized, a cell must express an antigen-specific receptor, either a B-cell receptor (BCR) or T-cell receptor (TCR). As a consequence of the tolerizing interaction, the cell or the individual exposed to the antigen is said to be tolerant. There are several stages of development of lymphocytes during which tolerance can be induced to provide protection against the development of autoimmunity. Tolerance induced during the early stages of development is referred to as central tolerance; tolerance induced in mature lymphocytes is referred to as peripheral tolerance. There are multiple mechanisms by which tolerance may be induced, and these are discussed further in the sections that follow.
Central tolerance occurs in the primary lymphoid organs (the bone marrow for B cells and the thymus for T cells) as part of the process known as negative selection (see Chapters 8 and 10). Negative selection in central lymphoid organs ensures that the majority of B and T cells that emerge in the periphery react only with foreign antigens. Nevertheless, this process is not perfect—some autoreactive cells inevitably escape to peripheral lymphoid organs such as the spleen and lymph nodes. If this happens, peripheral tolerance mechanisms may keep them under control.
How do lymphocytes distinguish self from nonself? As the experiments of Owen and Medawar’s group showed, self-antigens have no specific characteristics that distinguish them from foreign antigens. Thus, lymphocytes must rely on some other characteristics to make this distinction. The modern interpretation of Medawar’s studies of neonatal tolerance is that exposure of immature lymphocytes to antigen results in tolerance, whereas the first exposure of mature lymphocytes to the same antigen results in activation. What conditions favor the development of tolerance rather than activation? One is that immature lymphocytes are much more sensitive than mature lymphocytes to strong signals delivered through their antigen receptors. This is the basis for negative selection of B cells in the bone marrow and T cells in the thymus.
Another scenario that favors tolerance is continuous exposure of lymphocytes to high levels of antigen, which can lead to chronic antigen receptor signaling. Lymphocytes are continuously exposed to high levels of self-antigen, while exposure to foreign antigen is generally abrupt and short lived. Lymphocytes also learn to distinguish self from nonself by the context in which they encounter the antigen. If the antigen is encountered in the absence of co-stimulatory signals, then the lymphocytes are tolerized; if antigen is encountered in the presence of co-stimulatory signals, the lymphocytes are activated. Because the immune response is focused on responding to foreign antigens such as infectious organisms, foreign antigens are much more likely to come in contact with lymphocytes in the presence of co-stimulatory signals; thus, foreign antigens are more likely than self-antigens to activate lymphocytes.
Central Tolerance
The random process of generating diversity in BCRs and TCRs inevitably generates receptors that can recognize self-antigens. This concept was initially proposed in the 1950s by Sir MacFarlane Burnet in his clonal selection theory, for which he won the Nobel Prize in 1960. Burnet hypothesized that autoreactive lymphocytes—B and T cells with receptors for self-antigen—pose a danger to the host and must be purged or deleted from the developing lymphocyte repertoire to maintain tolerance to self. As we mentioned above, tolerance induced during the early stages of development is referred to as central tolerance. We now know that mechanisms other than deletion may be involved in central tolerance, but Burnet’s hypothesis is still substantially correct. The mechanisms of central tolerance and the conditions under which it is implemented are explored below.
Mechanisms of Central Tolerance: T and B Cells
Deletion.
Deletion is a mechanism by which autoreactive T and B cells are eliminated from the repertoire by undergoing apoptosis (programmed cell death). For developing T cells, deletion in the thymus is the major mechanism of negative selection—thymocytes expressing TCRs that interact with high affinity with self-antigens plus self-MHC (expressed on thymic dendritic cells and medullary epithelial cells) undergo rapid deletion (see Figure 13.1). This prevents mature autoreactive T cells from exiting the thymus and moving out into the periphery.
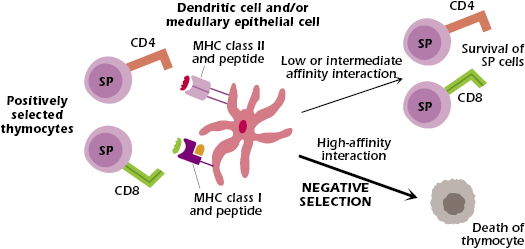
For a long time immunologists were puzzled as to how T cells specific for nonthymic, tissue-specific self-antigens, such as those expressed in the pancreas, could be eliminated in the thymus, since these self-antigens were not thought to be expressed in the thymus. The answer became apparent from studies over the past several years to uncover the cause of autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), a disease in which children suffer from a number of autoimmune symptoms including hypoparathyroidism, adrenal insufficiency, thyroiditis, type I diabetes mellitus, and ovarian failure. The studies showed that expression of the product of the AIRE (autoimmune regulator) gene is defective in patients with APECED (see Chapter 10). The AIRE gene product is a transcription factor regulator expressed predominantly in the thymus, by thymic medullary epithelial cells. The AIRE gene product facilitates the expression in the thymus of self-antigens that are normally found outside the thymus; for example, self-antigens expressed in the pancreas and central nervous system. Thus, in normal individuals, negative selection of self-reactive T cells specific for self-molecules expressed by peripheral tissues occurs. However, in APECED patients, and AIRE gene knockout mice, there is little expression in the thymus of these peripheral tissue self-antigens. The lack of negative selection permits autoreactive T cells to escape thymic deletion and exit to the periphery, where they may induce autoimmune disease.
Mechanisms of Central Tolerance: B Cells
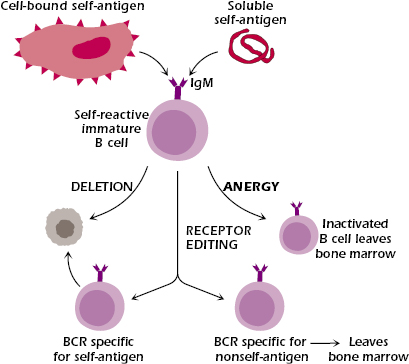
Central tolerance in the B-cell lineage shows some features that are also found in central tolerance in the T-cell lineage. In particular, negative selection—deletion of autoreactive B cells—occurs in the primary lymphoid organ; that is, the interaction in the bone marrow of self-antigen with an immature IgM+ B cells expressing a BCR specific for a self-antigen can result in deletion (shown in Figure 8.2 in Chapter 8). The best evidence for deletion of autoreactive B cells came from studies of transgenic mice. When mice transgenic for an antibody specific for the protein hen egg lysozyme (HEL) were bred with transgenic mice expressing HEL as a membrane-bound autoantigen, anti-HEL-specific B cells were deleted in the bone marrow when they came in contact with membrane HEL (see left side of Figure 13.2).
Central tolerance in B cells also shows some features that are not found in the development of central tolerance in T cells. These are described in the sections that follow.
Anergy.
Anergy is defined as the functional inactivation of a lymphocyte, resulting in unresponsiveness upon further contact with antigen. Anergic B cells cannot be activated to proliferate and differentiate into antibody-secreting cells upon engagement of their BCRs. Likewise, anergic T cells cannot be activated to proliferate and secrete cytokines following ligation of their TCR with antigen in the context of self-MHC.
Evidence for central B-cell anergy was first observed in 1980 by Sir Gustav Nossal and Beverly Pike. They demonstrated that when a cross-linking signal was delivered to immature B cells via their BCR, the B cells were anergized and were unable to mature or to undergo activation. B-cell anergy has since been observed in transgenic mouse systems. In the transgenic HEL model described in the section above, when transgenic mice making antibody specific for HEL were bred with transgenic mice expressing HEL as a soluble autoantigen, the offspring of these mice produced HEL-specific B cells that were not deleted after contact with antigen. These HEL-specific B cells were present but unable to respond to antigen; they had been rendered anergic or unresponsive (see right side of Figure 13.2). Experiments using mice transgenic for anti-DNA and other anti-self-antigens gave similar results, establishing that anergy is an important mechanism that prevents autoantibody-specific B cells from secreting autoantibodies.
The difference between the results of the two anti-HEL transgenic models was interpreted as suggesting that an encounter with a self-antigen that is membrane-bound induces extensive cross-linking of the BCR and delivers a strong signal that results in deletion; an encounter with a soluble self-antigen that induces more moderate cross-linking delivers a weaker signal that induces anergy. Thus, the degree of aggregation or polymerization of autoantigens appears to be a key factor in the mechanism of B-cell tolerance.
There is little evidence of anergy as a mechanism in the development of central tolerance for T cells in the thymus. Rather, as described above, most negative selection of autoreactive T cells in the thymus appears to occur by deletion. As described below though, anergy is a major mechanism in peripheral T-cell tolerance.
Receptor Editing.
Receptor editing is an important mechanism in developing B cells: it allows an immature B cell expressing a BCR specific for a self-antigen to replace it with a BCR specific for a nonself-antigen (see Figure 13.2 and Chapter 8). Receptor editing occurs predominantly in light chain genes: The existing rearranged light chain variable region is replaced with an upstream variable segment that rearranges to a more downstream J region. This secondary rearrangement does not always eliminate autoreactivity; that is, receptor editing may generate a BCR that is still self-reactive. In this case, rearrangement continues until all available upstream variable regions have been exhausted or until a non-autoreactive receptor is generated. If continued editing fails to result in a non-autoreactive BCR, the B cell undergoes deletion. Evidence for in vivo BCR editing was first observed in the bone marrow of mice transgenic for an antibody to an endogenous MHC class I molecule and in mice transgenic for an antibody to double-stranded DNA (dsDNA). Unlike B cells, developing T cells do not appear to use receptor editing as a means of altering their specificity.
Importance of receptor avidity in tolerance. What determines which of the spectrum of mechanisms leading to tolerance will govern the fate of selected immature lymphocytes? It is currently believed that the threshold of receptor signaling is the major force that determines which mechanism of tolerance governs immature lymphocytes. The strength, or avidity, of interaction of the BCR or TCR with its autoantigen influences cell fate. Avidity with respect to B cells depends on the affinity of interaction between the BCR and its antigen, density of the surface BCR, and the nature and concentration of the autoantigen. The higher the affinity of the antibody for its antigen and the more BCR expressed on the B-cell membrane, the greater the avidity of the antigen–antibody interaction. This correlates directly with the extent of immunoglobulin receptor cross-linking. The nature of the autoantigen also influences its ability to induce BCR cross-linking. For example, multivalent and membrane-bound antigens mediate more extensive receptor cross-linking than monovalent or soluble antigens. Initially, a B cell with a high avidity for an autoantigen will undergo receptor editing in an attempt to avert autoreactivity. B cells with more moderate avidity for an autoantigen are targeted to anergy, while B cells with a weak avidity for autoantigen become clonally ignorant.
In the thymus, the affinity of the TCR for self-peptide plus self-MHC, the level of TCR on the surface of the T cell, and the level of MHC molecules expressed on the surface of the APC determine avidity and affect T-cell selection. In addition, the type of cell presenting self-antigen to the T cell seems to influence whether the T cell undergoes positive or negative selection. T cells that recognize peptide plus MHC expressed on epithelial cells in the cortex are positively selected. At the corticomedullary junction, T cells come in contact with self-antigen and MHC molecules expressed on dendritic cells. It is in this context that T cells with a high avidity for peptide plus MHC undergo negative selection and are deleted. It is not known whether dendritic cells deliver signals different from cortical epithelial cells, and thus leading to negative rather than positive selection of self-reactive T cells. Anergy and editing seem to play minor roles in central T-cell tolerance.
Peripheral Tolerance
Occasionally, autoreactive B cells and T cells escape negative selection and enter the periphery. This can result from a self-antigen not being expressed at a high enough level in the primary lymphoid organ (bone marrow or thymus) or because the developing lymphocyte has a weak affinity for self-antigen. Such autoreactive lymphocytes generally do not pose a threat to the host—a state referred to as clonal ignorance. However, clonally ignorant lymphocytes may be activated if the concentration of autoantigen is increased, e.g., when large amounts of intracellular self-antigens are released (following cell death) or when a strong co-stimulatory signal is delivered to the clonally ignorant cell during an infection. In addition, B cells sometimes acquire autoreactive specificities through somatic mutation in the periphery.
Peripheral tolerance has evolved as a safety net to catch autoreactive B and T cells that escape to or arise in the periphery. Peripheral tolerance also prevents us from responding to every foreign antigen we encounter, such as those in our food. Mechanisms of peripheral tolerance include anergy, Fas-mediated activation-induced cell death, and the induction of regulatory T (Treg) cells. Although receptor editing has been shown to occur in peripheral B cells, it is not clear whether the role of editing in the periphery is to avert autoreactivity or to increase B-cell diversity.
Anergy
Peter Bretscher and Mel Cohn were the first to hypothesize that B cells specific for thymus-dependent (TD) antigens require two signals to be activated: the first signal provided by antigen, and the second signal by T cells. They also hypothesized that B cells would be rendered unresponsive in the absence of the second signal. These hypotheses are generally accepted. We now know that this second signal can be provided by the engagement of CD40 ligand (CD40L) on the surface of the T cell with CD40 expressed on the B cell. Studies in transgenic mice have confirmed that B cells exposed to self-antigen become anergized in the absence of T-cell help (Figure 13.3). Since autoreactive T cells usually get deleted in the thymus, they are generally not available in the periphery to provide co-stimulatory help to autoreactive B cells.
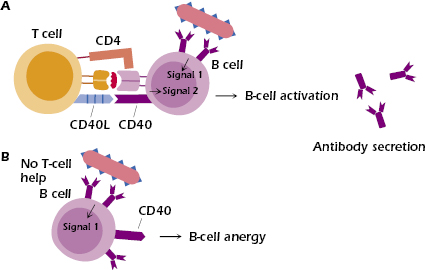
Anergic B cells cannot successfully compete for entry into B-cell follicles in the spleen and lymph nodes with nonanergic B cells. Instead they are arrested in development at the T cell–B cell border and soon die by apoptosis. It is now thought that follicular exclusion occurs because anergic B cells require higher concentrations of a cytokine known as BAFF (B-cell activating factor of the tumor necrosis factor family, also known as Blys) for survival than naïve B cells; in an environment with a limited amount of BAFF, anergic B cells are at a disadvantage and therefore undergo cell death instead of entering follicles.
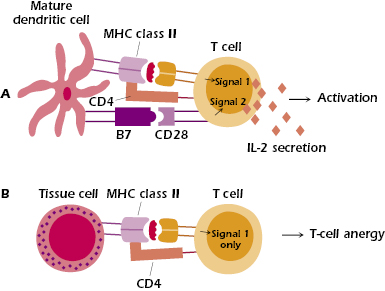
T-cell anergy is believed to be a major mechanism of tolerance to self-antigens that are encountered in the periphery. T cells also require two signals to be activated (see Figure 13.4 and Chapter 11). The first signal (Signal 1) is provided by the MHC–peptide complex, and the second signal (Signal 2) is provided by interactions between co-stimulatory molecules expressed on the antigen-presenting cell (APC) and the respective ligands on the T cell. A major co-stimulatory signal is provided by the ligation of B7-1 (CD80) and/or B7-2 (CD86) expressed on the APC with CD28 expressed on the T cell. Delivery of Signal 1 to the T cell leads to the induction of several transcription factors, one of which binds to the promoter region of the IL-2 gene, allowing for its transcription. If Signal 1 is not accompanied by the co-stimulatory B7–CD28 interaction, then IL-2 mRNA is rapidly degraded, IL-2 protein is not made, and the T cell fails to be activated. Tissue cells (such as liver cells) do not express co-stimulator molecules, and APCs do not constitutively express them: B cells, macrophages, and dendritic cells must be activated before they express co-stimulatory molecules (see Chapter 11). Hence, if a tissue cell (Figure 13.4) or unactivated APC presents antigen to a T cell, the T cell will be anergized because of the absence of a co-stimulatory signal.
Regulatory T Cells
In the early 1970s, experiments suggested that a specialized population of T cells—T suppressor cells—suppressed the responses of other lymphocytes. However, the inability to isolate and clone a suppressor T-cell population led to skepticism about whether this population really existed. Renewed interest in suppressor cells emerged in the 1990s with the identification of a population of CD4+ T cells with the ability to downregulate T-cell function. It was initially observed that neonatal thymectomy of mice resulted in autoimmunity, which could be prevented by transferring splenocytes from untreated mice back into the thymectomized mice. The suppressive activity in the splenocytes was eventually linked to a population of CD4+ T cells that became known as suppressor or natural regulatory T cells (Treg). These T cells expressed CD25, the IL-2 receptor α chain. Studies of immune dysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) in humans and of Scurfy disease in mice led to the identification of a marker specific for Treg, known as Foxp3 (forkhead box P3). Foxp3 is a transcription factor required for the development of Treg in the thymus, and has been shown to induce T cells to acquire regulatory activity in the periphery. Foxp3 serves as an intracellular marker for Treg. Mutations in the X-linked Foxp3 gene observed in IPEX patients and Scurfy mice lead to severe autoimmunity in affected males. A cell surface molecule known as GITR (glucocorticoid-induced TNF receptor family-related gene) has also been shown to be expressed at high levels on Treg cells.
As described in Chapters 10 and 11, natural CD4+CD25+ Treg cells represent a distinct lineage of T cells that is selected during T-cell development in the thymus. Treg cells constitute approximately 10% of peripheral CD4+ T cells. These cells are capable of dramatic expansion in cell numbers during immune responses. Global depletion of Treg cells in mice leads to organ-specific autoimmune diseases, such as thyroiditis, gastritis, and type I diabetes, which can be averted by Treg replacement. In addition to their role in self-tolerance, Treg can suppress graft versus host disease (discussed in Chapter 19); immune responses to tumor cells, allergens, and pathogens; and immune responses to organ transplants. Treg cells require IL-2 for their maintenance and activation, and have been shown to utilize a diverse repertoire of TCRs. They seem to have an increased affinity for self-peptide–MHC class II complexes, compared to helper T cells. Treg cells require specific TCR engagement to be activated, but once activated, their suppressive function is antigen nonspecific. Thus, Treg can suppress autoreactive effector CD4+ and CD8+ T cells that are specific for antigens different from those recognized by the Treg cells themselves. Treg cells have also been shown to suppress B cells, dendritic cells, and natural killer (NK) cells.
Many mechanisms have been proposed to explain the suppressive function of Treg cells. Figure 13.5 shows how Treg cells may inhibit responses of effector CD4+ T cells. Evidence suggests that CTLA-4, which is constitutively expressed on Treg cells, plays an important role in Treg-mediated suppression, since blockade of CTLA-4 results in organ-specific autoimmune disease. One hypothesis suggests that interaction of B7 (CD80 and CD86) on APCs with CTLA-4 on Treg cells delivers a co-stimulatory signal to the Treg that induces their suppressive activity. In addition, lymphocyte activation gene 3 (LAG3), an adhesion molecule expressed on the surface of Treg cells, binds MHC class II molecules and is thought to have a role in the suppressive activity of Treg cells. Upon engagement with MHC class II molecules, LAG3 expressed on Treg may induce downregulation of B7 on dendritic cells, making them weaker activators of effector T cells.
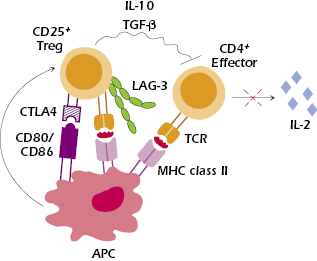
Suppression of responder T cells involves inhibiting their proliferation and activation and preventing their production of IL-2. Several studies suggest that IL-10 and TGF-β are needed for Treg suppression, but this remains controversial. Secretion of IL-10 by Treg cells in the lamina propria has been shown to control colitis by suppressing resident autoreactive T cells; however, blocking IL-10 in vitro fails to abrogate suppression. Involvement of cell surface expression of TGF-β by Treg cells has been reported in suppression mediated by cell contact. However, Treg cells isolated from mice deficient in TGF-β still have immunosuppressive activity. Thus, TGF-β may be more involved in the maintenance of Treg cells.
CD4+CD25+ Treg cells are also clinically relevant. Enhancing either the number or activity of these cells may be important for treating autoimmune diseases, and for suppression of allograft rejection. In addition, depletion of these T cells may enhance immune responses to tumor vaccines and vaccines for infectious agents such as human immunodeficiency virus (HIV). Other types of T-regulatory cells have been described in various experimental systems. When they are induced, they are referred to as adaptive T-regulatory cells. These may play a particularly important role in modulating immune reactivity to spontaneous inflammatory disease, such as inflammatory bowel disease; or in modifying immune responsiveness in infectious diseases, such as leishmaniasis. In many systems, cytokines such as IL-10 and TGF-β play an important role in mediating immune responsiveness.
Natural and adaptive regulatory T cells are involved in maintaining homeostasis between immune responsiveness and self-tolerance. The redundancy of these regulatory T-cell subsets (that is, how one subset may overlap in function with the others), their specialized roles in dampening the immune response, and their relationship to one another are areas of active research.
Fas–FasL Interactions
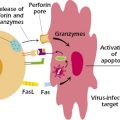
Fas-mediated apoptosis, shown in Figure 13.6, is thought to play a critical role in the removal of mature autoreactive B and T lymphocytes. Fas (CD95), expressed by activated lymphocytes and other cells, is a member of the tumor necrosis factor (TNF) receptor family. Fas binds to Fas ligand (FasL, CD178), expressed by several cell types, including activated T cells and certain epithelial cells. When FasL binds to Fas, it causes Fas to trimerize. The ligation of Fas initiates apoptosis in the cell expressing Fas, by activating a “death” domain in Fas. The activated Fas death domain interacts with the death domains of several cytosolic adaptor proteins, the most important of which is FADD (Fas-associated death domain). The interaction of Fas and FADD then triggers the binding of procaspase 8 and the subsequent activation of a cascade of caspases—cysteine proteases—that ultimately result in the apoptosis of the cell. T cells may express both Fas and FasL and so may be both a target and perpetrator of this mechanism of death, depending on the circumstances. Anergic B cells, some of which express Fas, are also susceptible to Fas-mediated apoptosis: Interaction with CD4+ T cells that express FasL leads to death of these self-reactive B cells.
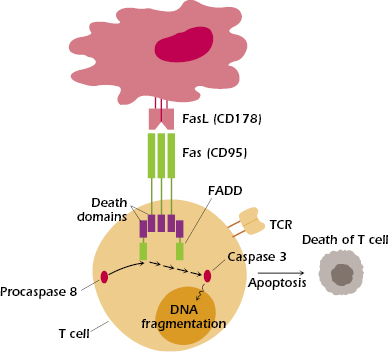
People with a mutated Fas gene develop an autoimmune condition known as autoimmune lymphoproliferative syndrome (ALPS), because autoreactive B and T lymphocytes are not properly deleted in the periphery. This results in the accumulation of lymphocytes and dramatic enlargement of lymph nodes and spleen. The importance of Fas–FasL interactions was initially established by studies of two mouse strains with an autoimmune condition that causes them to accumulate enormous numbers of lymphocytes in the spleen and lymph nodes. These mouse strains, known as lpr (for lymphoproliferative) and gld (for generalized lymphoproliferative disease), have mutations in the Fas and FasL genes, respectively. These mutations impair the deletion of autoreactive B and T cells.
Oral Tolerance
Oral tolerance is specific immune unresponsiveness to ingested food antigens. It is mediated by T cells, and the mechanisms for establishing it are dependent on the dose of ingested antigen. A low dose of ingested antigen seems to induce regulatory T cells, while a high dose seems to induce T-cell anergy or deletion. Oral tolerance is first initiated when orally administered antigen encounters gut-associated lymphoid tissue (GALT), which includes the epithelial cells of the intestinal villi, intraepithelial lymphocytes, lamina propria lymphocytes, and lymphoid nodules of the Peyer’s patches. Dendritic cells, the major APCs in the GALT, process and present most ingested antigens; however, other APCs, such as macrophages, B cells, and epithelial cells, also play a role. When a large dose of food antigen is administered, not all of it is degraded by the time it reaches the small intestine; consequently, some food antigens get absorbed intact into the systemic circulation. The antigen is then processed and presented by peripheral APCs in the absence of co-stimulatory interactions, which results in T-cell anergy.
Low-dose tolerance is induced locally, in the gut. The cytokine milieu of the GALT, which contains elevated levels of IL-4, IL-10, and TGF-β, promotes differentiation into TH2 and regulatory T cells and inhibits differentiation into TH1 cells. The regulatory T cells induced are specific for the ingested antigen, yet they mediate nonspecific suppression via secretion of TGF-β.
Low doses of oral antigen can suppress rodent models of autoimmune diseases, such as in experimental autoimmune encephalomyelitis (EAE, a model of multiple sclerosis), collagen-induced arthritis in susceptible strains, and autoimmune thyroiditis. Presumably the oral ingestion of antigen protects by the induction of regulatory T cells. Treatment of human autoimmune disease with oral antigens has been much less successful. One reason why oral antigen treatment of patients with autoimmune diseases has met with only limited success is that it is more difficult to suppress the function of T cells that are already activated. In the animal models, antigens can be administered before inducing disease and before T cells are activated. In addition, the cytokine environment in the human gut may differ from that of rodents’.
Immune Privilege
There are several sites in the body that do not develop immune responses to pathogens, tumor cells, or histoincompatible tissue transplants. These sites, known as immune privileged sites, include the eye, testis, brain, ovary, and placenta. The immune privilege of the eye is illustrated by the fact that corneal transplants in humans do not require tissue matching or immunosuppressive therapy. It was previously believed that immune protection in privileged sites was predominantly due to (1) lack of lymphatic drainage and (2) the blood barrier; both of these inhibit inflammatory cells from reaching antigens in the privileged sites. However, it is now known that other factors, such as the immunosuppressive cytokines IL-10 and TGF-β, and expression in privileged sites of FasL, play important roles in establishing immune privilege. Studies have demonstrated that FasL is expressed on several types of cells found in immune privileged sites; the interaction of these FasL-expressing cells with infiltrating Fas-expressing inflammatory T cells leads to apoptosis of the T cells. Human retinal pigment epithelial (RPE) cells and corneal endothelial cells have been observed to express FasL and to induce apoptosis of inflammatory T cells. The success of corneal transplants in mice has been shown to be due to expression of FasL by cells of the graft, which prevent inflammatory damage to the graft by eliminating infiltrating Fas+ T cells. When corneas from mice lacking functional FasL (gld mice) are transplanted into allogeneic recipients, the grafts are rejected because infiltrating Fas+ lymphocytes cannot be eliminated by FasL-mediated apoptosis. Human RPE cells have also been shown to secrete soluble factors such as TGF-β that suppress infiltrating T cells.
Autoimmunity and Disease
At the turn of the twentieth century, the German bacteriologist and immunologist Paul Ehrlich coined the term horror autotoxicus to describe a potentially toxic immune response to self. According to Ehrlich, the body was incapable of producing such an autoimmune response because of the “horror” this would inflict on the individual. We now know that autoreactive lymphocytes do arise, both centrally and peripherally, but they are normally kept in check and prevented from expanding by mechanisms of tolerance that we described earlier in the chapter. However, sometimes these tolerance mechanisms go awry and autoreactive lymphocytes escape regulatory checkpoints. If they are then triggered to undergo activation, autoimmunity may ensue.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


