After completion of this chapter, the reader will be able to: 1. Describe the prevalence of thrombotic disease in developed countries. 3. Distinguish between venous and arterial thrombosis. 4. Differentiate among acquired thrombosis risk factors related to lifestyle and disease, and congenital risk factors, noting which can be assessed in the hemostasis laboratory. 5. Discuss in relative terms, such as “most commonly affected” or “uncommonly affected,” the frequency with which various heritable risk factors occur in different ethnic groups. 6. Show how a hemostasis laboratory scientist may communicate with clinicians regarding the use of thrombosis risk profiles and screening tests. 7. List and employ the tests for predictors of arterial thrombotic disease, such as high-sensitivity C-reactive protein, homocysteine, fibrinogen, lipoprotein (a), and factor assays, to assess thrombotic risk. 8. Offer a sequence of lupus anticoagulant antibody tests that provides the greatest diagnostic validity and interpret the results of the tests. 9. Develop, carry out, and report the results of an antithrombin testing protocol. 10. List and employ tests of the protein C pathway, including protein C and protein S activity and concentration, activated protein C resistance, and the factor V Leiden assay, to assess thrombotic risk. 11. Explain the molecular test for prothrombin G20210A mutation and the relationship of this mutation to venous thrombosis, and interpret given test results. 12. Describe the causes and pathophysiology of disseminated intravascular coagulation (DIC). 13. List and perform the assays comprising a primary test profile for diagnosis and management of DIC in an acute care facility. 14. Discuss the value of quantitative D-dimer assays. 15. Describe the efficacy of the assays used to monitor localized thrombosis: prothrombin fragment 1+2, fibrinopeptide A, and thrombin-antithrombin complex. 16. Describe the cause and clinical significance of heparin-induced thrombocytopenia (HIT). 17. Describe the clinical diagnosis, laboratory diagnosis, and management of HIT. Before 1992, medical laboratory scientists performed assays to detect only three proven venous thrombosis risk factors: deficiencies of the coagulation control factors antithrombin, protein C, and protein S.1 Taken together, these three deficiencies accounted for no more than 7% of cases of recurrent venous thromboembolic disease and bore no relationship to arterial thrombosis. Since the report by Dahlback et al2 of activated protein C (APC) resistance in 1993 and the characterization by Bertina et al3 of the factor V Leiden (FVL) mutation as its cause in 1994, efforts devoted to thrombosis prediction and evaluation have redefined the hemostasis laboratory and increase its workload exponentially. The list of current assays includes APC resistance, FVL mutation, prothrombin G20210A mutation, factor VIII activity, lupus anticoagulant (LA), anticardiolipin (ACL) antibodies, and anti–β2-glycoprotein I (anti–β2-GPI) for venous thrombosis and tissue plasminogen activator (TPA), plasminogen activator inhibitor 1 (PAI-1), high-sensitivity C-reactive protein (CRP), lipoprotein (a), plasma homocysteine, and tests for platelet activity such as urinary 11-dehydrothromboxane B2 (see Chapters 44 and 46) for arterial thrombosis. Technical improvements have also been made in the diagnostic accuracy of integrated LA identification kits, the quantitative D-dimer assay, and tests for coagulation activation markers such as prothrombin fragment 1+2 (PF 1+2) and thrombin-antithrombin (TAT) complex.4 In addition, old tests, such as the fibrinogen assay and the factor VIII assay, have been applied to the prediction of venous and arterial thrombotic risk. Thrombosis is a multifaceted disorder resulting from abnormalities in blood flow, such as stasis, and abnormalities in the coagulation system, platelet function, leukocyte activation molecules, and the blood vessel wall. Thrombosis is the inappropriate formation of platelet or fibrin clots that obstruct blood vessels. These obstructions cause ischemia (loss of blood supply) and necrosis (tissue death).5 Thrombophilia is defined as the predisposition to thrombosis secondary to a congenital or acquired disorder. The theoretical causes of thrombophilia are the following: • Physical, chemical, or biologic events such as chronic or acute inflammation that release prothrombotic mediators from damaged blood vessels or suppress blood vessel production of normal antithrombotic substances • Inappropriate and uncontrolled platelet activation • Uncontrolled triggering of the plasma coagulation system At least 30% of the world’s people are expected to die of a thrombotic condition, and 25% of initial thrombotic events are fatal.6 Many fatal thromboses go undiagnosed before autopsy. The annual incidence of venous thrombosis in the unselected U.S. population is 1 in 1000.7,8 This includes the comparatively innocent thrombosis of superficial leg veins as well as the more dangerous deep vein thromboses that form in the iliac, popliteal, and femoral veins of the upper legs and calves.9 Large occlusive thrombi also may form, although less often, in the veins of the upper extremities, liver, spleen, intestines, brain, and kidneys. The symptoms of thrombosis include the sensation of heat, localized pain, redness, and swelling. In deep vein thrombosis, the entire leg swells. Fragments of thrombi, called emboli, may separate from the proximal end of a venous thrombus, move swiftly through the right chambers of the heart, and lodge in the arterial pulmonary vasculature, causing ischemia and death of lung tissue.10 Nearly 95% of these pulmonary emboli arise from thrombi in the deep leg and calf veins. Of the 250,000 individuals who experience pulmonary embolism in the United States annually, 10% to 15% die within 3 months. Many pulmonary emboli go undiagnosed because of the ambiguity of the symptoms. Coagulation system imbalances, such as inappropriate activation, gain of coagulation factor function, inadequate control, or fibrinolysis, are the mechanisms most often implicated in venous thrombosis.11 Cardiovascular disease causes 500,000 premature deaths annually in the United States; 500,000 cerebrovascular accidents (strokes) result in 100,000 stroke-related deaths. About 80% of myocardial infarctions and 85% of strokes are caused by thrombi that block coronary arteries or carotid end arteries of the vertebrobasilar system, respectively.12 Transient ischemic attacks and peripheral arterial occlusions are more frequent than strokes and coronary artery disease and, although not fatal, cause substantial morbidity. One important mechanism for arterial thrombosis is the well-described atherosclerotic plaque formation in the vessel walls. Activated platelets, monocytes, and macrophages embed the fatty plaque within the endothelial lining, suppressing the normal release of antithrombotic molecules such as nitric oxide and exposing prothrombotic substances such as tissue factor (see Chapter 40). Small unstable plaques rupture, occluding arteries and releasing thrombotic mediators to trigger thrombotic events. Activated platelets also form arterial platelet plugs with von Willebrand factor and only minimal fibrin, the “white thrombi” that cause death of surrounding tissue (see Chapter 13). In life, we acquire a legion of habits and conditions that either maintain or damage our hemostasis systems. Their multiplicity makes it difficult to pinpoint precisely the factors that contribute to thrombosis or to determine which have the greatest influence. These factors seem to contribute to venous and arterial thrombosis in varying degrees. Table 42-1 lists the nondisease risk factors implicated in thrombosis.13 TABLE 42-1 Nondisease Risk Factors That Contribute to Thrombotic Disease In addition to life events, several conditions and diseases threaten us with thrombosis. Some are listed in Table 42-2, with an indication of the laboratory’s diagnostic contribution.14 TABLE 42-2 Diseases with Thrombotic Risk Components DIC, Disseminated intravascular coagulation; Fg, fibrinogen. Malignancies often are implicated in venous thrombosis. One mechanism is tumor production of tissue factor analogues that trigger chronic low-grade disseminated intravascular coagulation (DIC). In addition, stasis and inflammatory effects increase the risk of thrombosis. Migratory thrombophlebitis, or Trousseau syndrome, is a sign of occult adenocarcinoma such as cancer of the pancreas or colon.15 Myeloproliferative neoplasms such as essential thrombocythemia and polycythemia vera (see Chapter 34) may trigger thrombosis, probably through platelet hyperactivity. A cardinal sign of acute promyelocytic leukemia (see Chapter 36) is DIC secondary to the release of procoagulant granules from the malignant promyelocytes. DIC can intensify during therapy at the time of vigorous cell lysis.16 Paroxysmal nocturnal hemoglobinuria (see Chapter 23) is caused by a stem cell mutation that modifies membrane-anchored platelet activation suppressors. Venous or arterial thromboses occur in at least 40% of cases.17 Chronic inflammatory diseases cause thrombosis through a variety of mechanisms, such as elevated fibrinogen and factor VIII, decreased fibrinolysis, promotion of atherosclerotic plaque formation, and reduced free protein S activity secondary to increased C4b-binding protein (C4bBP). Diabetes mellitus is a particularly dangerous chronic inflammatory condition, raising the risk of cardiovascular disease sixfold. Conditions associated with venous stasis, such as congestive heart failure, also are common risk factors for venous thrombosis. Untreated atrial fibrillation increases the risk of ischemic strokes due to clot formation in the right atrium and embolization to the brain.18 Nephrotic syndrome creates protein imbalances that lead to thrombosis through loss of plasma procoagulants. Nephrotic syndrome may also cause hemorrhage (see Chapter 41).19 Congenital thrombophilia is suspected when a thrombotic event occurs in young adults; occurs in unusual sites such as the mesenteric, renal, or axillary veins; is recurrent; or occurs in a patient who has a family history of the disorder (Table 42-3).20 Because thrombosis is multifactorial, however, even patients with congenital thrombophilia are most likely to experience thrombotic events because of a combination of constitutional and acquired conditions.21 TABLE 42-3 Predisposing Congenital Factors and Thrombosis Risk APC resistance is found in 3% to 8% of whites. Resistance extends to Arabs and Hispanics, but the mutation is absent from African and East Asian populations (Table 42-4).22 APC resistance may exist in the absence of the FVL mutation and occasionally is acquired in pregnancy or in association with oral contraceptive therapy. TABLE 42-4 The prothrombin G20210A gene mutation is the second most common inherited thrombophilic tendency in patients with a personal and family history of deep vein thrombosis.23 All together, protein C, protein S, and antithrombin deficiencies are found in 0.2% to 1.0% of the world population. The incidences of dysfibrinogenemia and the various forms of abnormal fibrinolysis (plasminogen deficiency, TPA deficiency, and PAI-1 excess) are under investigation. Thrombosis often is associated with a combination of genetic defect, disease, and lifestyle influences. The fact that an individual possesses protein C, protein S, or antithrombin deficiency does not mean that thrombosis is inevitable. Many heterozygotes experience no thrombotic event during their lifetimes, whereas others experience clotting only when two or more risk factors converge. A young woman heterozygous for the FVL mutation has a 35-fold increase in thrombosis risk upon starting oral contraceptive therapy. In the Physicians’ Health Study, homocysteinemia tripled the risk of idiopathic venous thrombosis, and the FVL mutation doubled it. When both were present, the risk of venous thrombosis was increased 10-fold.24 When thrombophilia is suspected, it is important to assess all known risk factors, because it is the combination of positive results that determine the patient’s cumulative risk of thrombosis.25 The presence or absence of laboratory-detected risk factors does not affect anticoagulant treatment, however, when thrombosis is in progress. Current anticoagulant therapy and ongoing or recent thrombotic events affect the interpretation of antithrombin, protein C, protein S, factor VIII, and LA testing. These assays should be performed 10 to 14 days after anticoagulant therapy is withdrawn (Table 42-5). TABLE 42-5 Complete Thrombophilia Laboratory Test Profile *Inaccurate during active thrombosis or anticoagulant therapy. Perform 14 days after anticoagulant therapy is discontinued. APL antibodies are a family of immunoglobulins that bind protein-phospholipid complexes.26 APL antibodies include LAs, detected by clot-based profiles, and ACL antibodies and anti–β2-GPI antibodies, detected by immunoassay. Chronic autoimmune APL antibodies are associated with APS, which is characterized by transient ischemic attacks, strokes, coronary and peripheral artery disease, venous thromboembolism, and repeated pregnancy complications.27,28 APL antibodies arise as immunoglobulin M (IgM), IgG, or IgA isotypes. Because they may bind a variety of protein-phospholipid complexes, they are called nonspecific inhibitors. Their name reflects the fact that they previously were thought to bind phospholipids directly; however, their target antigens are the proteins assembled on anionic phospholipid surfaces.29 The plasma protein most often bound by APL antibodies is β2-GPI, although annexin V and prothrombin are sometimes implicated. APL antibodies probably develop in response to newly formed protein-phospholipid complexes, and investigators still are learning exactly how they cause thrombosis.30,31 Between 1% and 2% of unselected individuals of both sexes and all races, and 5% to 15% of individuals with recurrent venous or arterial thrombotic disease have APL antibodies.32 Most APL antibodies arise in response to a bacterial, viral, fungal, or parasitic infection or to treatment with numerous drugs (Box 42-1) and disappear within 12 weeks. These are mostly transient alloimmune APL antibodies and have no clinical consequences.33 Nevertheless, the laboratory scientist must follow up any positive results on APL antibody assays to determine their persistence. Clinicians suspect APS in unexplained venous or arterial thrombosis, thrombocytopenia, or recurrent fetal loss.34 Specialized clinical hemostasis laboratories offer APL detection systems that include clot-based assays for LA and immunoassays for ACL and anti–β2-GPI antibodies. Occasionally, an LA is suspected because of an unexplained prolonged partial thromboplastin time (PTT) that does not correct when the test is repeated in mixing studies (see Chapter 45).35,36 Test systems that employ low-reagent phospholipid concentrations are sensitive to LA.37 There are four such systems, at least two of which are required to make up an LA profile. The need for multiple test systems arises from the multiplicity of LA reaction characteristics, and a confirmed positive result in one system is conclusive despite a negative result in another. The four available systems are PTTs formulated with low reagent phospholipid concentrations and silica-based particulate activators designed to be LA sensitive: the dilute Russell viper venom time (DRVVT), the kaolin clotting time (KCT), and the dilute thromboplastin time (DTT).38 As illustrated in Figure 42-1, the KCT and PTT trigger coagulation at the level of factor XII; DRVVT, at factor X; and DTT, at factor VII (see Chapter 45). The 2009 International Society on Thrombosis and Haemostasis (ISTH) update of guidelines for LA detection recommends using the DRVVT and LA-sensitive PPT as the two primary screening tests (Figures 42-2 and 42-3). This combination of assays updates the following 1995 ISTH requirements for LA identification39: 1. Prolonged phospholipid-dependent clot formation using a screen such as the low phospholipid PTT, DRVVT, KCT, or DTT 2. Failure to correct the prolonged clot formation by mixing with normal platelet-poor plasma and repeating the test (mixing study) 3. Shortening or correction of the prolonged screen test result by addition of excess phospholipids When the PTT or DRVVT is prolonged and anticoagulant therapy, particularly treatment with unfractionated heparin, has been ruled out, a new aliquot of patient platelet-poor plasma is mixed 1 : 1 with pooled normal platelet-poor plasma, and the assay is repeated on the mixture using the same test system (immediate mix). Some LAs are time and temperature dependent, so if the result for the mixture shows correction to within 10% or to within 5 seconds of the result for the normal platelet-poor plasma, the test should be repeated again after incubating the mix at 37° C for 1 to 2 hours. Each laboratory manager or director must decide what degree of shortening constitutes correction. Many use the Rosner index, which defines correction as a mixture result within 10% of the result of the platelet-poor normal plasma.40 Many others define correction as return to a value within the PTT reference interval. If either the PTT or the DRVVT remains prolonged (that is, uncorrected), the presence of LA is presumed, and confirmation is necessary. Confirmatory tests employ the same assay that originally yielded prolonged results, adding a high-concentration phospholipid reagent such as hexagonal phase phospholipid. If LA was the cause of prolongation, the high-phospholipid test result shortens (corrects).41 LA and ACL antibodies coexist in 60% of cases, and both may be found in APS.42 The ACL test is an immunoassay that may be normalized among laboratories and is not affected by heparin therapy, oral anticoagulant therapy, current thrombosis, or factor deficiencies. The manufacturer coats microplate wells with bovine heart cardiolipin and blocks (fills open receptor sites) with a bovine serum solution containing β2-GPI. Test sera or plasmas are serially diluted and pipetted to the wells along with calibrators and controls. ACL binds the solid-phase cardiolipin–β2-GPI target complex and cannot be washed from the wells. Enzyme-labeled anti–human IgG, IgM, or IgA conjugates are added. A color-producing substrate is then added, and a color change indicates the presence of ACL. Color intensities of the patient and control samples are compared with the calibrator curve. Results are expressed using GPL, MPL, or APL units, where 1 unit is equivalent to 1 mcg/mL of an affinity-purified standard IgG, IgM, or IgA specimen.43 Reference limits are established in each laboratory. IgM and IgG anti–β2-GPI immunoassays are performed as a part of the profile that includes ACL assays.44 An anti–β2-GPI result of greater than 20 GPL or MPL units correlates with thrombosis more closely than the presence of ACL antibodies. Any ACL or β2-GPI assay yielding positive results should be repeated on a new specimen collected after 12 weeks to distinguish a transient alloantibody from a chronic autoantibody (Figure 42-4). For cases in which an APL antibody is suspected but the routine LA, ACL, and β2-GPI assay results are negative, the clinician may wish to order the antiphosphatidylserine immunoassay to detect APL antibodies specific for phosphatidylserine.45 A result greater than or equal to 16 IgG or 22 IgM antiphosphatidylserine units is considered positive. The antiphosphatidylserine assay is available from specialty reference laboratories. Activated factors V and VIII (factors Va and VIIIa) are inactivated by the APC–protein S complex. A mutation in the factor V gene substitutes glutamine for arginine at position 506 of the factor V molecule (FV R506Q). The arginine is a cleavage site for APC, so the substitution slows or prevents hydrolysis of the factor V molecule (Figure 42-5). Resistant factor Va remains active and increases the production of thrombin. The factor V R506Q mutation is named for the city in the Netherlands in which it was first described, Leiden (FVL mutation). Between 3% and 8% of Northern European whites possess FVL (see Table 42-4).46 Owing to its prevalence and the associated 3-fold higher thrombosis risk (18-fold higher for homozygotes), the acute care hemostasis laboratory must provide APC resistance detection to screen for FVL.47 In the APC resistance clot-based assay, patient plasma is mixed 1 : 4 with factor V–depleted plasma.48 PTT reagent is added to two aliquots of the mixture and incubated for 3 minutes (Figure 42-6). A solution of calcium chloride is pipetted into one mixture, and the clot formation is timed. A solution of calcium chloride with APC is added to the second mixture and clotting is timed. The normal ratio of PTT results between the two assays is 1.8 or greater. In APC resistance, the ratio is less than 1.8.49
Thrombosis Risk Testing
Developments in Thrombosis Risk Testing
Introduction to Thrombosis
Etiology of Thrombosis
Prevalence of Thrombosis
Prevalence of Venous Thrombosis
Prevalence of Arterial Thrombosis
Thrombosis Risk Factors
Acquired Thrombosis Risk Factors
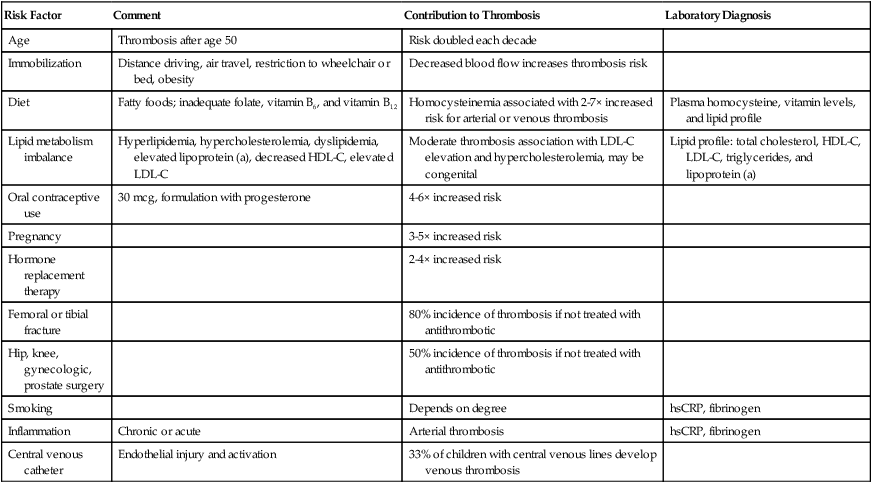
Risk Factor
Comment
Contribution to Thrombosis
Laboratory Diagnosis
Age
Thrombosis after age 50
Risk doubled each decade
Immobilization
Distance driving, air travel, restriction to wheelchair or bed, obesity
Decreased blood flow increases thrombosis risk
Diet
Fatty foods; inadequate folate, vitamin B6, and vitamin B12
Homocysteinemia associated with 2-7× increased risk for arterial or venous thrombosis
Plasma homocysteine, vitamin levels, and lipid profile
Lipid metabolism imbalance
Hyperlipidemia, hypercholesterolemia, dyslipidemia, elevated lipoprotein (a), decreased HDL-C, elevated LDL-C
Moderate thrombosis association with LDL-C elevation and hypercholesterolemia, may be congenital
Lipid profile: total cholesterol, HDL-C, LDL-C, triglycerides, and lipoprotein (a)
Oral contraceptive use
30 mcg, formulation with progesterone
4-6× increased risk
Pregnancy
3-5× increased risk
Hormone replacement therapy
2-4× increased risk
Femoral or tibial fracture
80% incidence of thrombosis if not treated with antithrombotic
Hip, knee, gynecologic, prostate surgery
50% incidence of thrombosis if not treated with antithrombotic
Smoking
Depends on degree
hsCRP, fibrinogen
Inflammation
Chronic or acute
Arterial thrombosis
hsCRP, fibrinogen
Central venous catheter
Endothelial injury and activation
33% of children with central venous lines develop venous thrombosis
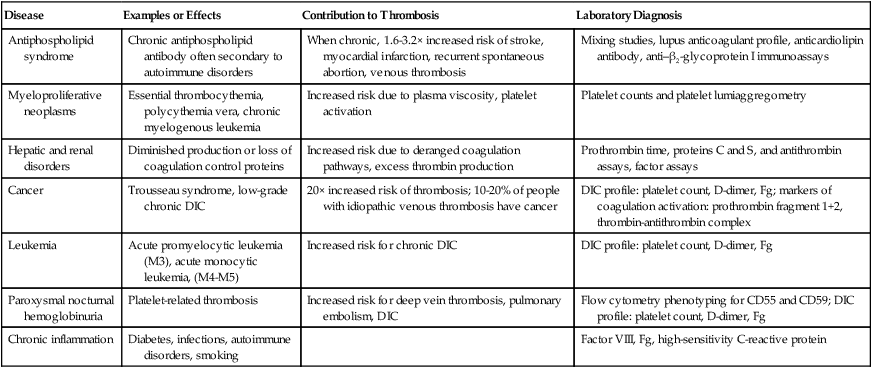
Thrombosis Risk Factors Associated with Systemic Diseases
Disease
Examples or Effects
Contribution to Thrombosis
Laboratory Diagnosis
Antiphospholipid syndrome
Chronic antiphospholipid antibody often secondary to autoimmune disorders
When chronic, 1.6-3.2× increased risk of stroke, myocardial infarction, recurrent spontaneous abortion, venous thrombosis
Mixing studies, lupus anticoagulant profile, anticardiolipin antibody, anti–β2-glycoprotein I immunoassays
Myeloproliferative neoplasms
Essential thrombocythemia, polycythemia vera, chronic myelogenous leukemia
Increased risk due to plasma viscosity, platelet activation
Platelet counts and platelet lumiaggregometry
Hepatic and renal disorders
Diminished production or loss of coagulation control proteins
Increased risk due to deranged coagulation pathways, excess thrombin production
Prothrombin time, proteins C and S, and antithrombin assays, factor assays
Cancer
Trousseau syndrome, low-grade chronic DIC
20× increased risk of thrombosis; 10-20% of people with idiopathic venous thrombosis have cancer
DIC profile: platelet count, D-dimer, Fg; markers of coagulation activation: prothrombin fragment 1+2, thrombin-antithrombin complex
Leukemia
Acute promyelocytic leukemia (M3), acute monocytic leukemia, (M4-M5)
Increased risk for chronic DIC
DIC profile: platelet count, D-dimer, Fg
Paroxysmal nocturnal hemoglobinuria
Platelet-related thrombosis
Increased risk for deep vein thrombosis, pulmonary embolism, DIC
Flow cytometry phenotyping for CD55 and CD59; DIC profile: platelet count, D-dimer, Fg
Chronic inflammation
Diabetes, infections, autoimmune disorders, smoking
Factor VIII, Fg, high-sensitivity C-reactive protein
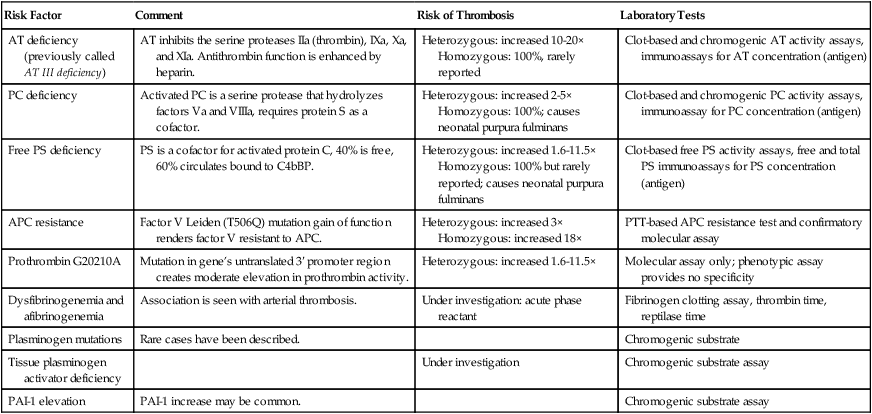
Congenital Thrombosis Risk Factors
Risk Factor
Comment
Risk of Thrombosis
Laboratory Tests
AT deficiency (previously called AT III deficiency)
AT inhibits the serine proteases IIa (thrombin), IXa, Xa, and XIa. Antithrombin function is enhanced by heparin.
Heterozygous: increased 10-20×
Homozygous: 100%, rarely reported
Clot-based and chromogenic AT activity assays, immunoassays for AT concentration (antigen)
PC deficiency
Activated PC is a serine protease that hydrolyzes factors Va and VIIIa, requires protein S as a cofactor.
Heterozygous: increased 2-5×
Homozygous: 100%; causes neonatal purpura fulminans
Clot-based and chromogenic PC activity assays, immunoassay for PC concentration (antigen)
Free PS deficiency
PS is a cofactor for activated protein C, 40% is free, 60% circulates bound to C4bBP.
Heterozygous: increased 1.6-11.5×
Homozygous: 100% but rarely reported; causes neonatal purpura fulminans
Clot-based free PS activity assays, free and total PS immunoassays for PS concentration (antigen)
APC resistance
Factor V Leiden (T506Q) mutation gain of function renders factor V resistant to APC.
Heterozygous: increased 3×
Homozygous: increased 18×
PTT-based APC resistance test and confirmatory molecular assay
Prothrombin G20210A
Mutation in gene’s untranslated 3′ promoter region creates moderate elevation in prothrombin activity.
Heterozygous: increased 1.6-11.5×
Molecular assay only; phenotypic assay provides no specificity
Dysfibrinogenemia and afibrinogenemia
Association is seen with arterial thrombosis.
Under investigation: acute phase reactant
Fibrinogen clotting assay, thrombin time, reptilase time
Plasminogen mutations
Rare cases have been described.
Chromogenic substrate
Tissue plasminogen activator deficiency
Under investigation
Chromogenic substrate assay
PAI-1 elevation
PAI-1 increase may be common.
Chromogenic substrate assay
Factor
Unselected Population
People with at Least One Thrombotic Event
Activated protein C resistance, factor V Leiden mutation
3-8% of whites, absent in Asians or Africans
20-25%
Prothrombin G20210A
2-3% of whites, absent in Asians or Africans
4-8%
Antithrombin deficiency
1 in 2000 to 1 in 5000
1-1.8%
Protein C deficiency
1 in 300
2.5-5.0%
Protein S deficiency
Unknown
2.8-5.0%
Hyperhomocysteinemia associated with methylenetetrahydrofolate reductase gene mutations
11%
13.1-26.7%
Dysfibrinogenemia
Unknown
1%
Abnormal fibrinolysis
Unknown
2%
Double Hit
Laboratory Evaluation of Thrombophilia
Assay
Reference Value/Interval
Comments
APC resistance
Ratio ≥1.8
Clot-based screen based on PTT and factor V–depleted plasma.
Factor V Leiden mutation
Wild-type
Molecular assay performed as follow-up to APC resistance ratio of <1.8
Prothrombin G20210A
Wild-type
Molecular assay. There is no phenotypic assay for prothrombin G20210A.
LA profile*
Negative for LA
Minimum of two clot-based assays such as PTT, DRVVT, KCT, or dilute PT with phospholipid neutralization test for immunoglobulins of APL family.
ACL antibody
IgG: <12 GPL
IgM: <10 MPL
Immunoassay for immunoglobulins of APL family. ACL depends on β2-GPI in reaction mix.
Anti–β2-GPI antibody
<20 G units
Immunoassay for an immunoglobulin of APL family. β2-GPI is key phospholipid-binding protein in family.
AT activity*
78-126%
Serine protease inhibitor suppresses IIa (thrombin), IXa, Xa, XIa. When consistently below reference limit, follow up with AT antigen assay.
PC activity*
70-140%
Digests VIIIa and Va. When consistently below reference limit, follow up with PC antigen assay.
PS activity*
65-140%
PC cofactor. When consistently below reference limit, follow up with total and free PS antigen assay, C4b-binding protein assay.
Factor VIII activity
50-186%
Confirm elevated factor VIII after 12 wk.
Fibrinogen
220-498 mg/dL
Clot-based assay. Elevation may be associated with arterial thrombosis.
Plasminogen activator inhibitor 1
<30 units/mL
Elevation may be associated with venous thrombosis.
Antiphospholipid Antibodies
Clinical Consequences of Antiphospholipid Antibodies
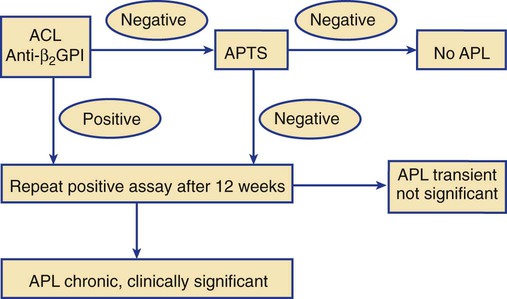
Detection and Confirmation of Antiphospholipid Antibodies
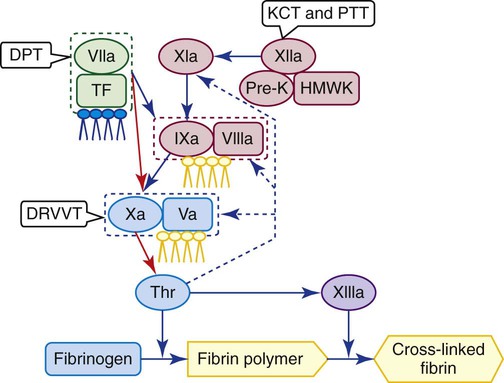
Lupus Anticoagulant Test Profile
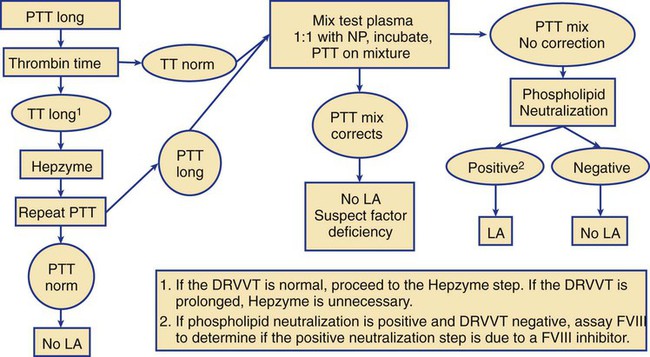
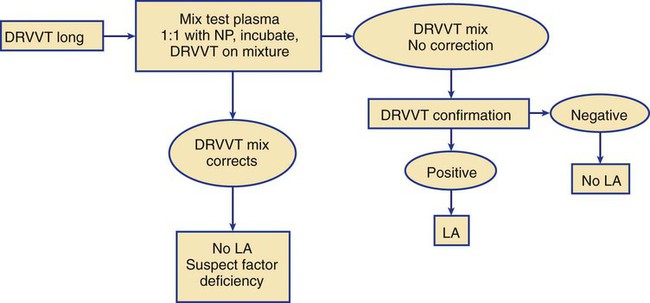
Performing the Clot-Based Lupus Anticoagulant Mixing Study
Anticardiolipin Antibody Immunoassay
Anti–β2-Glycoprotein I Immunoassay
Antiphosphatidylserine Immunoassay
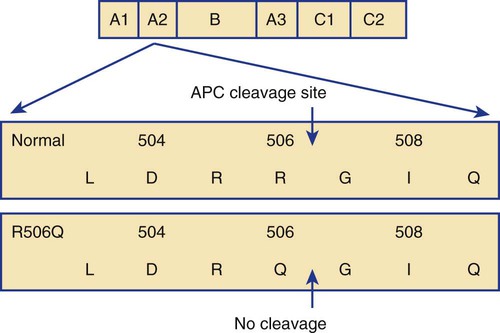
Activated Protein C Resistance and Factor V Leiden Mutation
Clinical Importance of Activated Protein C Resistance
Activated Protein C Resistance Clot-Based Assay
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thrombosis Risk Testing