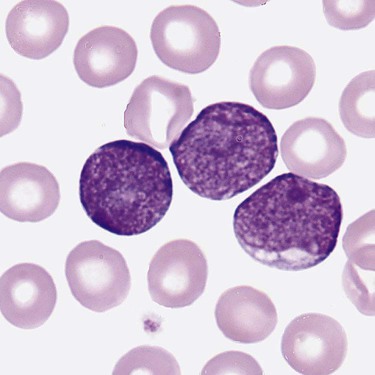
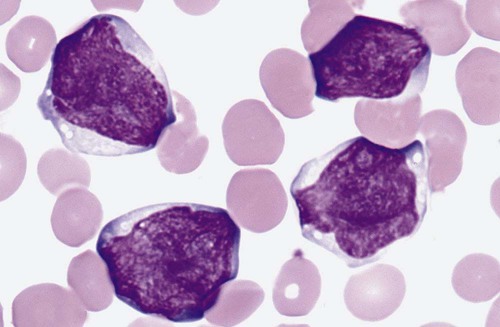
After completion of this chapter, the reader will be able to: 1. Clinically distinguish between acute lymphoblastic and acute myeloid leukemias. 2. Characterize the diagnostic criteria used for these leukemias. 3. Compare and contrast acute lymphoblastic and myeloid leukemias by morphology, presenting signs and symptoms, laboratory findings, and prognosis. 4. Interpret the results of diagnostic tests for acute leukemias. 5. Discuss tumor lysis syndrome, including risk, cause, and laboratory findings. 6. Discuss the rationale for the 2008 World Health Organization classification of acute leukemias. A 5-year-old child was seen by her family physician because of weakness and headaches. She had been in good health except for the usual communicable diseases of childhood. Physical examination revealed a pale, listless child with multiple bruises. The WBC count was 15 × 109/L, the hemoglobin was 8 g/dL, and the platelet count was 90 × 109/L. She had “abnormal cells” in her peripheral blood (Figure 36-1). Cytogenetic studies revealed hyperdiploidy. 1. What is the most likely diagnosis? 2. What characteristics of this disease indicate a positive prognosis? Acute lymphoblastic leukemia (ALL) is primarily a disease of childhood and adolescence, accounting for 25% of all childhood cancers and up to 75% of childhood leukemias.1 Although ALL is rare in adults, risk increases with age; most adult patients are older than 65 years.2 Treatment of childhood ALL is a triumph of modern hematology. A uniformly fatal disease before 1970, “good prognosis” ALL in children is associated with a 95% remission rate, and over 80% of patients are cured.1,3,4 The subtype of ALL is an important prognostic indicator for survival.5 Adults have a poorer outlook: 80% to 90% experience complete remission, but the cure rate is less than 40%.6,7 Only half of patients with ALL have leukocytosis, and many do not have circulating lymphoblasts.8 Neutropenia, thrombocytopenia, and anemia are usually present, which leads to fatigue (caused by anemia), fever (caused by neutropenia and infection), and mucocutaneous bleeding (caused by thrombocytopenia). Lymphadenopathy, including enlargement, is often a symptom.2 Enlargement of the spleen (splenomegaly) and of the liver (hepatomegaly) may be seen. Bone pain often results from infiltration of leukemic cells into the bone covering (periosteum).2 Eventual infiltration of malignant cells into the meninges, testes, or ovaries occurs frequently, and lymphoblasts can be found in the cerebrospinal fluid.9 In T-cell ALL (T-ALL), there may be a mass in the mediastinum, the area in the central chest containing the heart and its supporting nerves and vessels, thymus and other lymph nodes, trachea, esophagus, and so forth.10 The precursor lymphoid neoplasms are classified by the WHO into two major groups: B lymphoblastic leukemia/lymphoma and T lymphoblastic leukemia/lymphoma.11 B lymphoblastic leukemia/lymphoma is subdivided into seven subtypes that are associated with recurrent cytogenetic abnormalities. These entities are linked with unique clinical, phenotypic, or prognostic features (Box 36-1). Cases of B-cell ALL (B-ALL) that do not exhibit the specific genetic abnormalities are classified as B lymphoblastic leukemia/lymphoma, not otherwise specified. Although 50% to 70% of patients with T lymphoblastic leukemia/lymphoma have abnormal karyotypes, none of the abnormalities is clearly associated with specific biologic features, and thus T-ALL is not further subdivided.11 Lymphoblasts vary in size but fall into two morphologic types. The most common type seen is a small lymphoblast (1.0 to 2.5 times the size of a normal lymphocyte) with scant blue cytoplasm and indistinct nucleoli (see Figure 36-1); the second type of lymphoblast is larger (2 to 3 times the size of a lymphocyte) with prominent nucleoli and nuclear membrane irregularities10 (Figure 36-2). These cells may be confused with the blasts of acute myeloid leukemia (AML). Although morphology is the first tool used to distinguish ALL from AML, immunophenotyping (see Chapter 33) and genetic analysis (see Chapter 31) are the most reliable indicators of a cell’s origin. Because both B and T cells are derived from lymphoid progenitors, both express CD34, terminal deoxynucleotidyl transferase, and HLA-DR. Four types of ALL have been identified by immunologic methods: early B-ALL (progenitor B, pro-B, or pre-pre-B), intermediate (common) ALL, precursor B (pre-B) ALL, and T-ALL. B-ALL is characterized by specific B-cell antigens that are expressed at different stages of B-cell development. In general B cells express CD19, CD20, CD22, CD24, C79a, CD10, cytoplasmic µ, and PAX-5 (B-cell specific activator protein). The degree of differentiation of B-lineage lymphoblasts often correlates with genetics and plays an important role in treatment decisions.8,10,12 In the earliest stage of differentiation (pre-pre B or pro-B), blasts express CD19, cytoplasmic CD79a, cytoplasmic CD22, and nuclear TdT. The incidence of pro-B ALL is about 5% in children and 11% in adults. In intermediate B-ALL, also called common ALL, CD10 is expressed. CD10+ ALL is sometimes called common ALLa (cALLa), because this group comprises 65% of childhood ALL and 51% of adult ALL. In the most mature of the B-ALLs, called pre-B, cytoplasmic µ is present. Pre-B ALL accounts for 15% of childhood cases and 10% of adult B-ALL.13 T-ALL is seen most often in teenaged males with a mediastinal mass, elevated peripheral blast counts, meningeal involvement, and infiltration of extra marrow sites.14 The common T-cell markers CD2, CD4, CD5, and CD8 are usually present. In addition, cells may express CD3, CD1a, CD5, CD4, and CD8. Although this immunophenotype classically has been associated with a poor outcome, aggressive chemotherapy has resulted in improved prognosis; however, patients with T-ALL are more likely to relapse sooner than patients with B-ALL, and CNS relapse is more common.15 T-ALL accounts for about 1% of childhood cases and 27% of adult cases.13 Prognosis in ALL has improved dramatically over the past decade as a result of improvement in algorithms for treatment.10,16 The prognosis for ALL depends on age at the time of diagnosis, lymphoblast load (tumor burden), immunophenotype, and genetic abnormalities. Children rather than infants or teens do the best. Chromosomal translocations are the strongest predictor of adverse treatment outcomes for children and adults. Peripheral blood lymphoblast counts greater than 20 to 30 × 109/L, hepatosplenomegaly, and lymphadenopathy all are associated with worse outcome. In addition, the presence of an aberrant myeloid surface marker found in ALL (such as CD66c) is associated with a poor outcome in adults.17 The effects of other variables previously associated with a poorer prognosis, such as sex and ethnic group, have been eliminated when patients have been given equal access to treatment in trials carried out at a single institution.18 Cytogenetic abnormalities are seen in the majority of B- and T-cell ALL. In T-ALL there is less specificity and less correlation with treatment outcome than in B-ALL. B lymphoblastic leukemia/lymphoma with the t(9;22)(q34;q11.2);BCR-ABL1 mutation (Philadelphia chromosome–positive ALL), has the worst prognosis among ALLs. It is more common in adults (25% of all ALL cases) than in children (2% to 4% of ALL cases). Imatinib, which has shown success in treating chronic myelogenous leukemia, has improved survival (see Chapter 34). B lymphoblastic leukemia/lymphoma with t(v;11q23);MLL rearranged is more common in very young infants, and the translocation may even occur in utero.19 This leukemia has a very poor prognosis. About 25% of childhood ALL cases show a t(12;21)(p13;q22);ETV6-RUNX1 translocation and appear to derive from a B-cell progenitor, rather than the hematopoietic stem cell.20 This translocation is rare in adults. In children, it carries a very favorable prognosis, with a cure rate of over 90%. Hyperdiploidy in B lymphoblastic leukemia/lymphoma is common in childhood B-ALL, accounting for 25% of cases, but is much less common in adults. This genotype is associated with a very favorable prognosis in children, but a poor prognosis in adults.21 Conversely, hypodiploidy (less than 46 chromosomes) conveys a poor prognosis in both children and adults. B lymphoblastic leukemia/lymphoma with t(5:14)(q31;q32);IL3-IGH occurs in both children and adults, but is very rare, accounting for fewer than 1% of ALL cases. There are so few cases that the implication for prognosis is uncertain. B lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3);E2A-PBX1(TCF3-PBX1) accounts for about 6% of B-ALL cases in children. It is less common in adult ALL. Newer therapies have improved the outcome.8 At the morphologic and immunophenotypic levels, ALLs appear to be somewhat homogeneous, but at the genetic level, ALLs comprise a heterogeneous group.7,10 Thus, a variety of treatments are used. Patients first are classified into low-, standard-, and high-risk groups so that treatment can be appropriately directed. In general, children between the ages of 1 and 10 years with a white blood cell (WBC) count of less than 50 × 109/L, no extramedullary disease, and rapid response to initial therapy are classified into the low-risk group. High-risk features include age younger than 1 year, extramedullary spread, and poor response to initial therapy. Genetic features as detailed previously are also considered. Adults, by virtue of their age, are considered to be in the high-risk group.2 In general, treatment modalities include remission induction therapy, followed by consolidation (intensification), and then continuation therapy to eliminate residual disease. Central nervous system therapy (intrathecal route) is often included. Children with ALL may experience some paralysis or ability regression as a result of intrathecal therapy, in which chemotherapeutic drugs are administered directly into the central nervous system via the cerebrospinal fluid.22 Methotrexate, commonly used to treat ALL, is a folate antagonist that causes megaloblastosis on blood film or bone marrow smear (see Chapter 20) and rapid severe leukocytopenia and thrombocytopenia. Medications used for the treatment of adult ALL include daunorubicin, vincristine, and prednisone. Because daunorubicin frequently causes cardiac complications, routine cardiac enzyme assays are required. Vincristine attacks microtubule formation, is toxic to the nervous system, and interferes with platelet activity. Newer agents such as monoclonal antibodies (e.g., anti-CD52) are assuming the role of lead therapy, alone or in combination with more traditional chemotherapeutic drugs.23 Peripheral blood stem cell transplantation is the last line of treatment for unresponsive or relapsed ALL. (See Chapter 29 for more information on the treatment of WBC neoplasia.) AML is the most common family of leukemias in children younger than 1 year of age, but is rare in older children and adolescents. It is the most common type of leukemia in adults, and the incidence increases with age. The French-American-British (FAB) classification of AML was based on morphology and cytochemistry; the WHO classification relies heavily on cytogenetics and molecular characterization.8,11 The clinical presentation of AML is nonspecific but reflects decreased production of normal bone marrow elements. Most patients with AML have a total WBC count between 5 and 30 × 109/L, although the WBC count may range from 1 to 200 × 109/L. Myeloblasts are present in the peripheral blood in 90% of patients. Anemia, thrombocytopenia, and neutropenia give rise to the clinical findings of pallor, fatigue, fever with infections, bruising, and bleeding. In addition, disseminated intravascular coagulation and other bleeding abnormalities can be significant.24 Infiltration of malignant cells into the gums and other mucosal sites and skin also can be seen. Bone and joint pain are the first symptoms in only 25% of patients, unlike in ALL, in which such pain is common.25 Splenomegaly is seen in half of AML patients, but lymph node enlargement is rare. Cerebrospinal fluid involvement in AML is rare and does not seem to be as ominous a sign as in ALL. Patients with AML tend to have few symptoms related to the central nervous system, even when it is infiltrated by blasts. Common abnormalities in laboratory test results include hyperuricemia (caused by increased cellular turnover), hyperphosphatemia (due to cell lysis), and hypocalcemia (the latter two are also involved in progressive bone destruction). Hypokalemia is also common at presentation. During induction chemotherapy, especially when the WBC is quite elevated, tumor lysis syndrome may occur. Tumor lysis syndrome is a group of metabolic complications that can occur in patients with malignancy, most notably lymphomas and leukemias, with and without treatment of the malignancy.26 These complications are caused by the breakdown products of dying cancer cells, which in turn cause acute uric acid nephropathy and renal failure. Tumor lysis syndrome is characterized by hyperkalemia, hyperphosphatemia, hyperuricemia and hyperuricosuria, and hypocalcemia.25,26 The hyperkalemia alone can be life threatening. Aggressive prophylactic measures to prevent or reduce the clinical manifestations of tumor lysis syndrome are critical.27,28 The 2001 WHO classification incorporated genetic abnormalities into the classification of AML, and four categories of AML were identified. Since that time it has been recognized that not only genetic lesions recognizable at the microscopic level but also submicroscopic lesions cooperate to influence leukemogenesis, and seven subtypes of AML are included in the 2008 WHO classification.29 Laboratory diagnosis begins with a complete blood count, peripheral blood film examination, and bone marrow aspirate and biopsy specimen examination. The total WBC count is variable and may be normal, increased, or decreased; anemia is usually present, along with significant thrombocytopenia. The bone marrow is usually hypercellular, and greater than 20% of cells typically are marrow blasts, although if certain genetic abnormalities are present the 20% blast threshold is not necessary for the diagnosis of AML.8 Each category is discussed, and a summary of the classification is presented in Box 36-2.
Acute Leukemias
Case Study
Acute Lymphoblastic Leukemia
World Health Organization Classification
Morphology
Immunophenotyping
Prognosis
Genetics
Types of Treatment
Acute Myeloid Leukemia
Clinical Presentation
Subtypes of Acute Myeloid Leukemia and Related Precursor Neoplasms
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Acute Leukemias