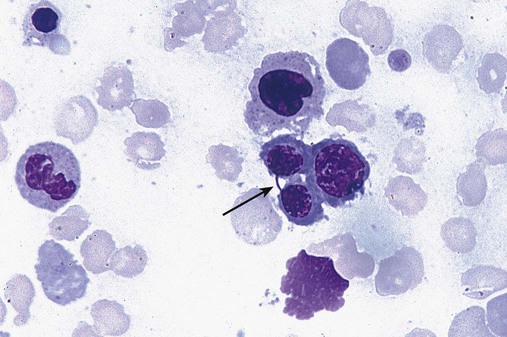
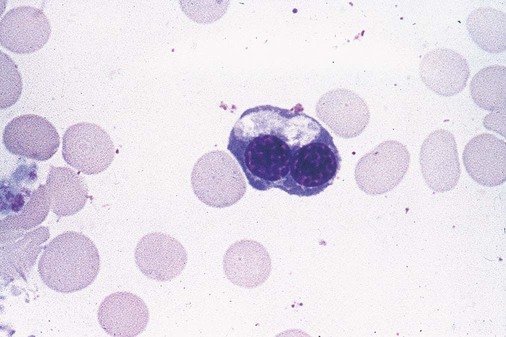
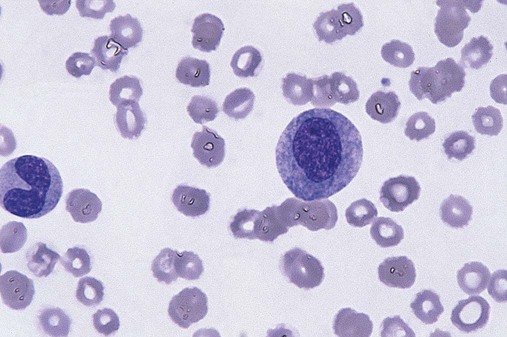
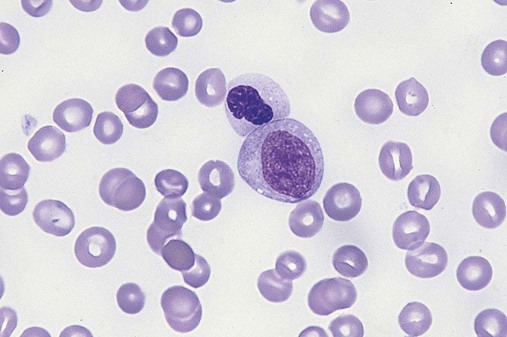
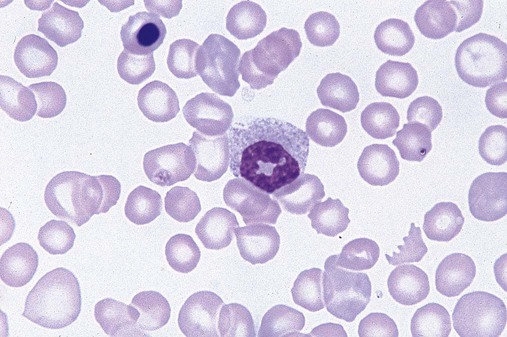
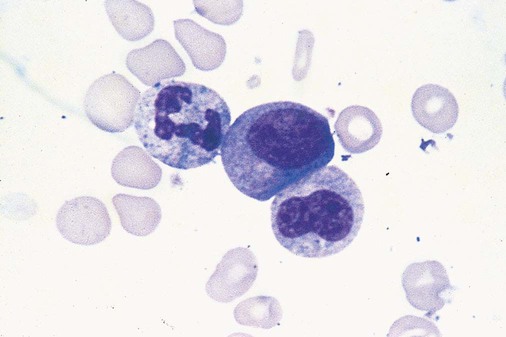
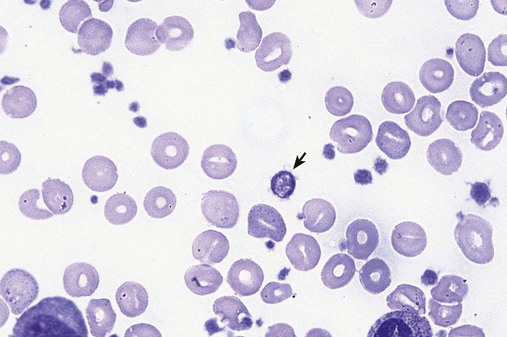
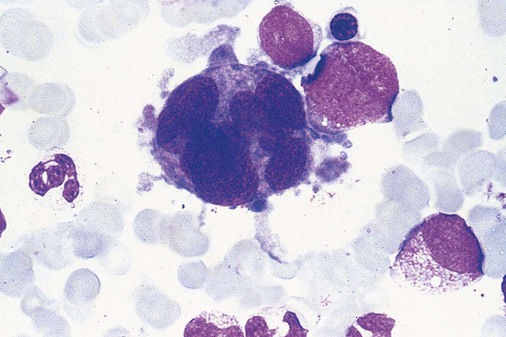
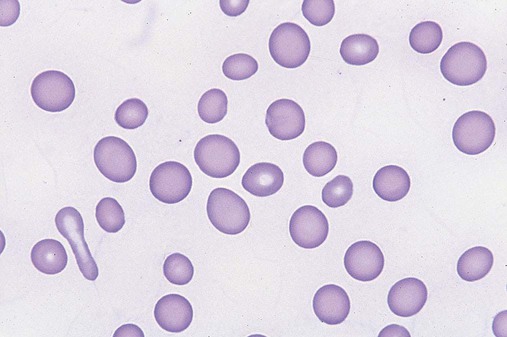
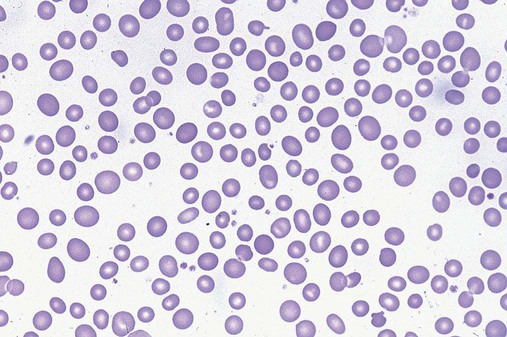
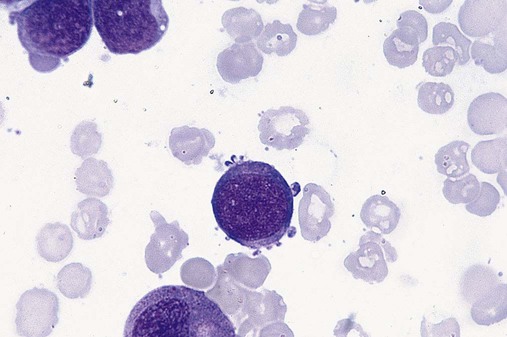
After completion of this chapter, the reader will be able to: 1. Define myelodysplastic syndromes (MDSs). 2. Explain the etiology of MDS. 3. Recognize morphologic features of dyspoiesis in bone marrow and peripheral blood. 4. Discuss abnormal functions of granulocytes, erythrocytes, and thrombocytes in MDS. 5. Correlate peripheral blood, bone marrow, and cytogenetic findings in MDS with classification systems. 6. Compare and contrast the French-American-British and the 2008 World Health Organization classifications of MDS. 7. Discuss prognostic indicators in MDS. 8. Discuss modes of management for MDS. 9. Discuss the epidemiology of MDS and apply it as a contributor in differential diagnosis. 10. Suggest laboratory tests and their results that would rule out MDS in the differential diagnosis. 11. Explain the rationale for the category of myelodysplastic/myeloproliferative neoplasms (MDS/MPN). 12. Correlate peripheral blood, bone marrow, and cytogenetic findings in MDS/MPN with disease classification. Historically, this pattern of abnormalities was referred to as refractory anemia, smoldering leukemia, oligoblastic leukemia, or preleukemia.1–3 In 1982, the French-American-British (FAB) Cooperative Leukemia Study Group proposed terminology and a specific set of morphologic criteria to describe what are now known as myelodysplastic syndromes (MDSs).4 In 1997, a group from the World Health Organization (WHO) proposed a new classification that included molecular, cytogenetic, and immunologic criteria in addition to morphologic features.5,6 The WHO classification was revised in 2008. Both the FAB and WHO classifications are discussed in this chapter. MDSs are a group of acquired clonal hematologic disorders characterized by progressive cytopenias in the peripheral blood, reflecting defects in erythroid, myeloid, and/or megakaryocytic maturation.7,8 The median age at diagnosis is 70. MDS rarely affect individuals younger than age 50 unless preceded by chemotherapy or radiation for another malignancy.1,9 Cases in young adults and children have been reported, however.10,11 The incidence of these disorders seems to be increasing, but this apparent increase may be attributable in part to improved techniques for identifying these diseases and to improved classification.12,13 At this time, the fastest-growing segment of the population is the group older than 60 years of age. MDSs are becoming a more common finding in the hematology laboratory, and familiarity with these disorders is an essential part of the body of knowledge of all medical laboratory professionals. MDS may arise de novo (primary MDS) or as a result of therapy (therapy-related MDS). Although MDSs are a group of heterogeneous diseases, all are the result of proliferation of abnormal stem cells.7,8,14,15 The initiating defect in most cases is at the level of the myeloid stem cell, because primarily the erythroid, myeloid, and megakaryocytic cells are affected. It may be that the affected hematopoietic stem cell has lost its lymphopoietic potential, because only rarely does MDS transform to acute lymphoblastic leukemia.16,17 The abnormal stem cell may be the result of the cumulative effects of environmental exposure in susceptible individuals. Mutations may be caused by chemical insult, radiation, or viral infection. There also may be an association with smoking.18 An association with inherited hematologic disorders has also be found.19 The mutated stem cell produces a pathologic clone of cells that expands in size at the expense of normal cell production.18,20 Because each mutation produces a unique clone with a specific cellular defect, MDSs have a multitude of expressions. Two morphologic findings are common to all types of MDS, however: the presence of progressive cytopenias despite cellular bone marrows and dyspoiesis in one or more cell lines. Disruption of apoptosis may be responsible for the ineffective hematopoiesis in MDS.21–26 Apoptosis (programmed cell death) regulates cell population by decreasing cell survival. In MDS, apoptosis is increased in early disease, when peripheral blood cytopenias are evident. Later in MDS, when progression toward leukemia is apparent, apoptosis has been shown to be decreased, which allows increased neoplastic cell survival and expansion of the abnormal clone.27–30 Other important factors include the levels of antiangiogenic cytokines, tumor necrosis factor, and cellular components of the immune system, as well as the interaction between MDS clonal cells and the hematopoietic inductive microenvironment. Evidence has shown that patients with MDS have increased levels of angiogenic growth factors, including vascular endothelial growth factor.31,32 Therapy-related MDS occurs in patients who have been treated previously with chemotherapy or radiotherapy or both. Median onset of therapy-related MDS varies with the agents used and is usually 2.5 to 5.0 years after therapy was initiated,33,34 although cases occurring 7 years after initial exposure to chemotherapy or radiation have been reported.35 Therapy-related MDS often is more aggressive and may evolve quickly into acute myeloblastic leukemia (AML).20,34,35 The 2008 WHO classification places therapy-related MDSs into the AML category of therapy-related myeloid neoplasms (see Chapter 36). In MDS each of the three major myeloid cell lines has dyspoietic morphologic features. The following sections provide descriptions of common abnormal morphologic findings.4,9,19 These descriptions are not all inclusive because of the large number of possible cellular mutations and combinations of mutations. In the peripheral blood, the most common morphologic finding in dyserythropoiesis is the presence of oval macrocytes (Figure 35-1). When these cells are seen in the presence of normal vitamin B12 and folate values, MDS should be included in the differential diagnosis. Hypochromic microcytes in the presence of adequate iron stores also are seen in MDS. A dimorphic red blood cell (RBC) population (Figure 35-2) is another indication of the clonality of this disease. Poikilocytosis, basophilic stippling, Howell-Jolly bodies, and siderocytes also are indications that the erythrocyte has undergone abnormal development.36 Dyserythropoiesis in the bone marrow is evidenced by RBC precursors with more than one nucleus or abnormal nuclear shapes. The normally round nucleus may have lobes or buds. Nuclear fragments may be present in the cytoplasm (Figure 35-3). Internuclear bridging is occasionally present (Figure 35-4).37 Abnormal cytoplasmic features may include basophilic stippling or heterogeneous staining (Figure 35-5). Ringed sideroblasts are a common finding. Megaloblastoid cellular development in the presence of normal vitamin B12 and folate values is another indication of MDS. The bone marrows in these cases may have erythrocytic hyperplasia or hypoplasia (Box 35-1). Dysmyelopoiesis in the peripheral blood is suspected when there is a persistence of basophilia in the cytoplasm of otherwise mature white blood cells (WBCs), indicating nuclear-cytoplasmic asynchrony (Figure 35-6). Abnormal granulation of the cytoplasm of neutrophils, in the form of larger than normal granules, hypogranulation, or the absence of granules, is a common finding. Agranular bands can be easily misclassified as monocytes (Figure 35-7). Abnormal nuclear features may include hyposegmentation, hypersegmentation, or nuclear rings (Figure 35-8).38 In the bone marrow, dysmyelopoiesis may be represented by nuclear-cytoplasmic asynchrony. Cytoplasmic changes include uneven staining, such as a dense ring of basophilia around the periphery with a clear unstained area around the nucleus or whole sections of cytoplasm unstained with the remainder of the cytoplasm stained normally (Figure 35-9). There may be abnormal granulation of the cytoplasm in which promyelocytes or myelocytes or both are devoid of primary granules (Figure 35-10), primary granules may be larger than normal, or secondary granules may be reduced in number or absent, and there may be an occasional Auer rod.39,40 Agranular promyelocytes may be mistaken for blasts; this could lead to misclassification of the disease in the AML scheme. Abnormal nuclear findings may include hypersegmentation or hyposegmentation and possibly ring-shaped nuclei (Box 35-2). Abnormal localization of immature precursors is a characteristic finding in bone marrow biopsy specimens from patients with MDS.41 Normally, myeloblasts and promyelocytes reside along the endosteal surface of the bone marrow. In some cases of MDS, these cells tend to cluster centrally in marrow sections. Platelets also exhibit dyspoietic morphology in the peripheral blood. Common changes include giant platelets and abnormal platelet granulation, either hypogranulation or agranulation (Figure 35-11). Some platelets may possess large fused granules. Circulating micromegakaryocytes may be present in peripheral blood from patients with MDS (Figure 35-12).9 The megakaryocytic component of the bone marrow may exhibit abnormal morphology: large mononuclear megakaryocytes, micromegakaryocytes, or micromegakaryoblasts. The nuclei in these cells may be bilobed or have multiple small, separated nuclei (Figure 35-13; Box 35-3).9 Dysplasia by itself is not sufficient evidence for MDS, because several other conditions can cause similar morphologic features. Some examples are vitamin B12 or folate deficiency, which can cause pancytopenia and dysplasia, and exposure to heavy metals. Copper deficiency may cause reversible myelodysplasia.42 Some congenital hematologic disorders, such as Fanconi anemia and congenital dyserythropoietic anemia, may also present with dysplasia. Parvovirus B19 and some chemotherapeutic agents may give rise to dysplasia similar to that in MDS. Paroxysmal nocturnal hemoglobinuria has similar features. Therefore a thorough history and physical examination, including questioning about exposure to drugs and chemicals, is essential.9 The cells produced by abnormal maturation not only have an abnormal appearance but also have abnormal function.9,43 The granulocytes may have decreased adhesion,44,45 deficient phagocytosis,45 decreased chemotaxis,44,45 or impaired microbicidal capacity.46 Decreased levels of myeloperoxidase and alkaline phosphatase may be found.47 The RBCs may exhibit shortened survival,48 and erythroid precursors may have a decreased response to erythropoietin that may contribute to anemia.49 Patients may experience increased bleeding despite adequate platelet numbers.9,50,51 The type and degree of dysfunction depend on the mutation present in the hematopoietic stem cell. In an effort to standardize the diagnosis of MDSs, the FAB created five classes of MDS, each with a specific set of morphologic criteria. The categories were defined by the amount of dysplasia and the number of blasts in the bone marrow. The diagnosis of acute leukemia required at least 30% blasts in the bone marrow.4 The FAB classification included the following: 2. Refractory anemia with ringed sideroblasts (RARS) 3. Refractory anemia with excess blasts (RAEB) 4. Chronic myelomonocytic leukemia (CMML) 5. Refractory anemia with excess blasts in transformation (RAEB-t) The FAB classification provided a framework for discussion of a seemingly heterogeneous group of disorders; however, its reliance on morphology alone limited its usefulness as a prognostic indicator. In addition, the FAB classification did not view MDSs in their totality, because it did not address therapy-related or hereditary forms, and the special case of childhood MDS was not considered. Advances in medical knowledge, including molecular analysis, have allowed integration of clinical, immunologic, genetic, and molecular data with morphologic features. The WHO classification retains many of the FAB features while recognizing molecular, cytogenetic, and immunologic characteristics of these disorders. The WHO classification also removed the problematic categories of CMML and RAEB-t and placed them in MDS/MPD and acute leukemia, respectively.19,52 The original modifications from the FAB classification of MDS included a reduction in the percentage of blasts required for diagnosis of AML from 30% to 20% and the recognition of two new classifications: refractory cytopenia with multilineage dysplasia (RCMD) and del(5q) syndrome. The 2008 revision of the WHO criteria added the category of refractory cytopenia with unilineage dysplasia (RCUD), refined some categories, and added the provisional category of childhood MDS, also called refractory cytopenia of childhood. The 2008 WHO classification is outlined in Box 35-4 and detailed in Table 35-1. The classification is extensive, and only the highlights are presented in this chapter.19
Myelodysplastic Syndromes
Etiology
Morphologic Abnormalities in Peripheral Blood and Bone Marrow
Dyserythropoiesis
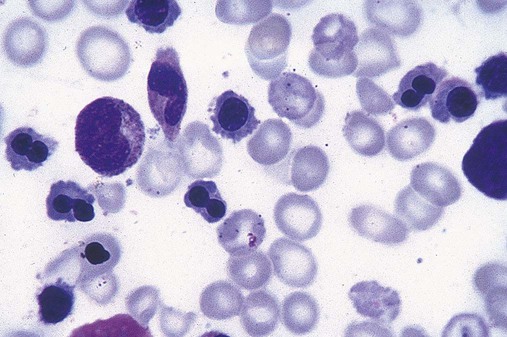
Dysmyelopoiesis
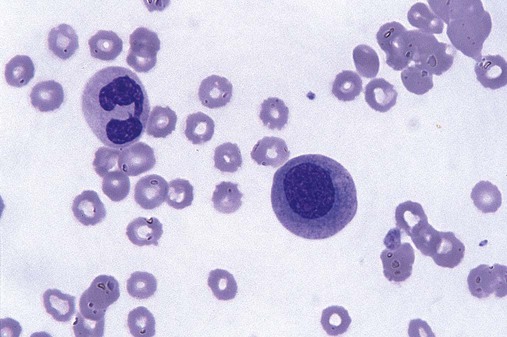
Dysmegakaryopoiesis
Differential Diagnosis
Abnormal Cellular Function
Classification of Myelodysplastic Syndromes
French-American-British Classification
World Health Organization Classification
Myelodysplastic Syndromes