Evolution of Therapeutic Approaches
The deadly novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), spread like wildfire across the entire world in the first exceptionally severe pandemic of the 21st century. Humanity had last seen a comparable worldwide viral infection in the 20th century, with the 1918 influenza pandemic caused by an H1N1 virus that infected approximately one-third of the world’s population; deaths were estimated at approximately 50 million people. ,
In this century, the first cases of a “pneumonia of unknown cause” were reported in Wuhan, China in December 2019, with the first death reported on January 11, 2020. A few days later, a dashboard showing COVID-19 cases in real time was started as a collaboration of several centers at Johns Hopkins University. The infection spread at such speed that the World Health Organization (WHO) declared it first to be a public health emergency and finally a pandemic on March 11, 2020, noting it was the first one caused by a coronavirus.
At that point, the number of cases outside China had increased 13-fold and the number of countries reporting cases had tripled. Dr. Tedros A. Ghebreyesus, WHO Director-General, expressed being deeply concerned “both by the alarming levels of spread and severity, and by the alarming levels of inaction,” and urged countries to take action immediately to contain the virus. The WHO recommended that people with mild respiratory symptoms should isolate themselves, and social distancing was advised even for countries with no reported cases. On March 11, 2020, there were 118,000 cases globally (in 114 countries) with 4291 deaths (case fatality rate [CFR] of 3.6%), of which 1267 cases and 38 deaths (CFR 3%) were in the United States.
Simultaneously, a review in JAMA Insights of the first published COVID-19 case studies from China reported that critical care would be “an integral component of the global response” to the infection. Approximately two-thirds of patients had been reported to require intensive care for respiratory support, after progressing to acute respiratory distress syndrome (ARDS). ARDS is characterized by (1) loss of integrity of capillary endothelium, (2) inflammation with recruitment of resident tissue macrophages, and (3) immune activation. Left untreated, ARDS can lead to pulmonary edema and death from progressive respiratory failure. The time between symptom onset and admission to the intensive care unit (ICU) was estimated to be 9 to 10 days. The authors raised the concern that in some regions, the lack of intensive care infrastructure might lead to health systems being overwhelmed if mechanical ventilation capacity were to be exceeded.
As patients with COVID-19 pneumonia were observed to develop ARDS, they were initially managed following widely accepted clinical practice guidelines, based on disease severity. Therapeutic measures for patients with ARDS included conservative fluid strategies for patients without shock after initial resuscitation, antibiotics for potential bacterial pneumonia or secondary infections, and lung-protective ventilation strategies such as mechanical ventilation with lower tidal volumes and lower inspiratory pressures. Proning for more than 12 hours per day was recommended for patients with severe ARDS, and disease management decisions were to be personalized for each patient. Modifications to the usual standard of care such as private rooms, distancing between patients, and exercising caution because of the risk for dispersion of aerosolized virus were implemented.
A case report of the clinical management of the first US COVID-19 case, diagnosed on January 20, 2020, was published in early March in the New England Journal of Medicine. The patient’s initial symptoms on admission were treated with antipyretics, fluid replacement, guaifenesin for cough, and antiemetics (ondansetron). As the disease progressed to pneumonia on day 9 of illness, the patient was treated with oxygen supplementation, antibiotics (vancomycin and cefepime), and compassionate use of an investigational antiviral therapy (intravenous remdesivir, an experimental antiviral).
Multiple therapies were tested during 2020 and 2021, in a desperate attempt to treat patients in a vacuum of definitive efficacy data. The US Food and Drug Administration (FDA) launched the Coronavirus Treatment Acceleration Program (CTAP), setting the regulatory stage for drug and biologicals manufacturers to develop products to meet this urgent need. As of May 12, 2021, excluding vaccines, there were more than 610 programs in planning stages, and more than 450 clinical trials had been reviewed by the FDA, resulting in nine COVID-19 treatments authorized for emergency use, and one treatment was approved ( Fig. 16.1 ).

In the face of the globally raging pandemic, the scientific community raced to unravel new antiviral therapies against COVID-19 disease. Meanwhile, they also, not surprisingly, explored the use of several existing antiviral, antimalarial, antiinflammatory, and even antibiotic agents for activity against the SAR-CoV-2 virus. One such repurposed drug is remdesivir, which was previously evaluated for treatment of Ebola virus disease and would become one of the most used tools in the COVID therapeutic armamentarium. A significant amount of research has been dedicated to remdesivir in COVID-19, as evidenced by the number of review articles published on this topic. Its definitive efficacy was tested in a randomized clinical trial (RCT) setting within the Adaptive COVID-19 Treatment Trial (ACTT), launched by the National Institute of Allergy and Infectious Diseases (NIAID) to evaluate the safety and efficacy of investigational therapeutics for treatment of severe COVID-19 in hospitalized adults. Remdesivir was found to be superior to placebo in shortening the time to recovery in adults hospitalized with COVID-19 and lower respiratory tract infection. Although hydroxychloroquine was the first repurposed medicine to receive an emergency use authorization (EUA) from the FDA, it was soon revoked because of lack of efficacy and potential safety concerns. , Remdesivir was the next to receive an EUA and the only therapy to date to have received full FDA approval ( Fig. 16.2 ).
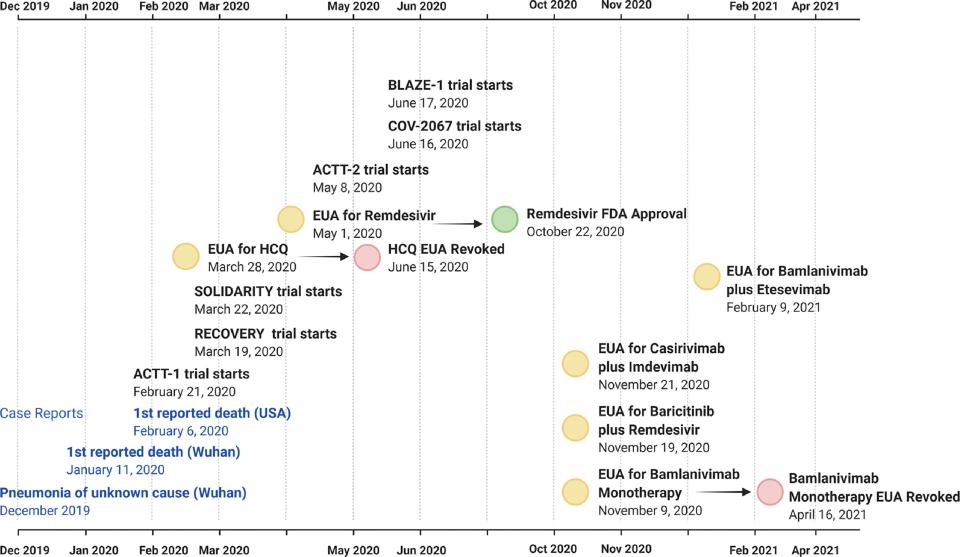
Similar to efforts in the United States, RCTs (SOLIDARITY and RECOVERY) were also initiated by the WHO and United Kingdom, respectively, to assess the efficacy and safety of a number of medicines previously approved for other uses. , Disappointingly, the SOLIDARITY trial reported little or no effect for remdesivir, hydroxychloroquine, lopinavir, or interferon regimens on overall mortality, initiation of ventilation, or duration of hospital stay in hospitalized patients with COVID-19. The RECOVERY trial investigated whether treatment with lopinavir-ritonavir, hydroxychloroquine, corticosteroids, azithromycin, colchicine, intravenous immunoglobulin (children only), convalescent plasma, synthetic neutralizing antibodies (REGN-COV2), tocilizumab, aspirin, baricitinib, infliximab, or anakinra (in children only) prevented death in patients with COVID-19. In this trial, encouraging results were observed with dexamethasone, which lowered the 28-day mortality among those who were receiving either invasive mechanical ventilation or oxygen alone at randomization. Also encouraging was the observation that tocilizumab, an interleukin-6 (IL-6) receptor monoclonal antibody, improved survival and other clinical outcomes in hospitalized COVID-19 patients with hypoxia and systemic inflammation, in addition to the benefits of systemic corticosteroids.
The adaptive design of the ACTT platform allowed for the evaluation of additional therapies over time, such as baricitinib, a Janus kinase (JAK) inhibitor, in combination with remdesivir in ACTT-2, and interferon-beta 1a (IFN-β1a) in combination with remdesivir, in ACTT-3. Baricitinib given in combination with remdesivir showed a shorter recovery time relative to remdesivir alone and led to an EUA from the FDA.
In parallel, neutralizing monoclonal antibodies (mAbs) that bind directly to the receptor binding domain (RBD) of the SARS-CoV-2 S protein to compete with the cellular receptor angiotensin-converting enzyme-2 (ACE2) emerged as promising therapeutic approaches for the treatment of COVID-19. Bamlanivimab monotherapy was the first mAb to receive an EUA for the treatment of mild to moderate COVID-19 in adults and pediatric patients at high risk for progressing to severe COVID-19 and/or hospitalization. However, this EUA was later revoked because of the potential for increased risk for treatment failure as a result of the emergence of SARS-CoV-2 viral variants resistant to bamlanivimab alone. It was later combined with another mAb, etesevimab, which binds to another distinct, but overlapping, epitope within the RBD of the S protein of SARS-CoV-2. The combination received an EUA based on the results of the BLAZE-1 trial. Another combination of RBD-binding mAbs, casirivimab and imdevimab, was also given an EUA based on the results of the COV-2067 trial.
In the following section, the therapies will be discussed in further detail. Although not widely authorized for emergency use, there are numerous other repurposed or repositioned drugs that have been evaluated in RCTs and are being used in varying degrees worldwide and thus are discussed in a separate section. This section also includes a comprehensive list of investigational therapies (new and repurposed) currently in clinical trials to serve as a reference point. Finally, the evolving treatment guidelines for COVID-19 management at different phases of the disease are discussed.
Treatments Authorized for Emergency Use
As part of the mechanism to facilitate availability of treatments, vaccines, and other measures during public health emergencies, the US FDA issued EUAs to a number of medicines as of May 15, 2021. The initial date of EUA issuance, authorized use, and primary endpoint data from the pivotal trial supporting the respective EUAs are presented in Table 16.1 .
Antiviral Monotherapy: Remdesivir
On May 1, 2020, the FDA issued an EUA for Veklury (remdesivir) for the treatment of hospitalized patients with severe COVID-19, defined as patients meeting one or more of the following four criteria ― (1) oxygen saturation (SpO 2 ), (2) requiring supplemental oxygen, (3) requiring mechanical ventilation, and (4) requiring extracorporeal membrane oxygenation (ECMO). The EUA was based on a phase III, randomized double-blind trial, the ACTT-1, in which treatment with remdesivir was compared with placebo. With supportive evidence from three randomized controlled clinical trials that included patients hospitalized with mild-to-severe COVID-19 (including ACTT), on October 22, 2020, a full approval was granted for the use of remdesivir (as Veklury) for the treatment of COVID-19 in adults and pediatric patients (12 years and older and weighing at least 40 kg), requiring hospitalization. It was the first treatment to gain full FDA approval for COVID-19.
Pharmacological Properties
Remdesivir (GS-5734) is a phosphoramidate prodrug of a nucleoside analog, GS-443902, that directly inhibits viral replication of SARS-CoV-2. It undergoes intracellular activation to form the pharmacologically active remdesivir triphosphate (RDV-TP), which competes with adenosine triphosphate (ATP) for incorporation into RNA-dependent RNA polymerase. This, in turn, leads to premature termination of viral RNA transcription and inhibition of subsequent RNA synthesis ( Fig. 16.3 ).

In cellular assays, remdesivir demonstrated antiviral activity against a clinical isolate of SARS-CoV-2 in primary human airway epithelial cells with a 50% effective concentration (EC 50 ) of 9.9 nM after 48 hours of treatment. In other cell lines (Calu-3 and A549-hACE2), EC 50 values were 115 nM and 280 nM after 48 and 72 hours, respectively.
Whereas remdesivir (the prodrug) and its metabolites (GS-704277 and GS-441524) can be measured in plasma, the active moiety (triphosphate GS-443902) can only be detected intracellularly. Therefore activation to the triphosphate form was evaluated using peripheral blood mononuclear cells as clinical surrogates. The pharmacokinetics of remdesivir and its metabolites (GS-704277 and GS-441524) was dose-proportional across the 3- to 225-mg dose range in healthy participants, after administration of single intravenous doses. Remdesivir was developed as an intravenous formulation because of its large first-pass hepatic extraction. After multiple once-daily doses of 150 mg remdesivir for 14 days, the major metabolite GS-441524 accumulated approximately 1.9-fold in plasma.
Dosing and Indication
Single-dose vials of remdesivir are available in both solution and lyophilized injection formulations; the lyophilized formulation has a long shelf life and is stable at room temperature because of its better physiochemical stability. The United States Prescribing Information (USPI) states that remdesivir is indicated for adults and pediatric patients (12 years of age and older and weighing at least 40 kg) for the treatment of COVID-19 requiring hospitalization, and that it should be administered only in a hospital or in a health care setting capable of providing acute care comparable to inpatient hospital care. The recommended dosage consists of a single intravenous loading dose of 200 mg remdesivir on day 1 followed by once-daily maintenance doses of 100 mg remdesivir starting on day 2. Before initiating remdesivir, estimated glomerular filtration rate (eGFR), prothrombin time, and liver function are assessed for any dosage adjustments. The recommended treatment duration is 5 days, which can be extended for up to 5 additional days (for a total treatment duration of up to 10 days) if no clinical improvement is observed.
Clinical Efficacy
NIAID ACTT-1 (in Mild/Moderate and Severe COVID-19): Primary Evidence of Efficacy
In this pivotal phase III trial designated ACTT-1 (NCT04280705), 1062 hospitalized patients with COVID-19 received a 10-day course of remdesivir or placebo. The primary outcome was the time to recovery by day 29, on which a patient met the criteria for category 1 (not hospitalized and no limitations of activities), 2 (not hospitalized, with limitation of activities, home oxygen requirement, or both), or 3 (hospitalized, not requiring supplemental oxygen and no longer requiring ongoing medical care) on an 8-category ordinal scale. The time to recovery was shorter for patients who received remdesivir compared with those on placebo (see Table 16.1 ). In the severe disease stratum (N = 957), the median time to recovery was 11 days, relative to 18 days (rate ratio [RR] for recovery, 1.31; 95% confidence interval [CI], 1.12–1.52). Mortality by day 29 was numerically lower in the remdesivir group (11.4%) compared with placebo (15.2%), but the difference was not statistically significant (hazard ratio [HR] 0.73; 95% CI, 0.52–1.03]).
GS-US-540-5773 (in Severe COVID-19): Supportive Data
This randomized, open-label trial (NCT04292899) evaluated the safety and efficacy of 5 days versus 10 days of remdesivir in hospitalized patients with severe COVID-19. The primary endpoint, clinical status on day 14, did not demonstrate a significant difference in efficacy between 5- and 10-day regimens of remdesivir. The absence of a standard-of-care alone arm limited the interpretability of the data. However, an external (synthetic) control arm based on retrospectively collected data from contemporaneously hospitalized patients with severe COVID-19 showed that by day 14, remdesivir was associated with 62% reduced odds of death (adjusted odds ratio [aOR], 0.38; 95% CI, 0.22–0.68, P = .001) versus standard-of-care treatment in patients with severe COVID-19.
GS-US-540-5774 (in Moderate COVID-19): Supportive Data
The randomized, open-label trial NCT04292730 evaluated the safety and efficacy of 5 days versus 10 days of remdesivir compared with standard-of-care in hospitalized patients with moderate COVID-19. A statistically significant difference in the odds of improvement at day 11 favoring the 5-day (but not the 10-day) treatment group over standard of care was demonstrated. Notwithstanding the limitations of its open-label design, this trial provided supportive evidence for the efficacy of remdesivir in patients hospitalized with COVID-19 of moderate severity (i.e., patients hospitalized but not requiring supplemental oxygen).
Clinical Safety
The safety profile of remdesivir was informed by data from three phase III studies in hospitalized adult patients with COVID-19 (N = 1313), four phase I studies in healthy adults (N = 131), and from patients with COVID-19 who received remdesivir under the EUA or a compassionate use program. In the comparative ACTT-1 trial, the rates of severe (grade 3) or potentially life-threatening (grade 4) adverse events for remdesivir were comparable to placebo (8% vs. 9%), as were serious adverse events (0.4% vs. 0.6%) and adverse events leading to study drug discontinuation (2% vs. 3%). Adverse events of lower grade (1 or 2) were not collected. The most common adverse reactions (≥5%, all grades) observed with remdesivir were nausea and elevations in liver transaminases. The overall efficacy and safety data support a flexible recommendation for 5- to 10-day treatment duration regimens to allow providers latitude in tailoring treatments per clinical response.
Key warnings and precautions include hypersensitivity reactions, which may be mitigated with slower infusion rates, and transaminase elevations requiring close laboratory testing and monitoring. Remdesivir is contraindicated in patients with a history of clinically significant hypersensitivity reactions, such as infusion-related and anaphylactic reactions. Coadministration of remdesivir with chloroquine or hydroxychloroquine is not recommended because of potential antagonistic effects on intracellular metabolic activation and the antiviral activity of remdesivir.
Immunomodulator and Antiviral Combination: Baricitinib and Remdesivir
On November 19, 2020 the FDA issued an EUA for the emergency use of baricitinib in combination with remdesivir for the treatment of COVID-19 (suspected or confirmed) in hospitalized adults and pediatric patients 2 years of age or older requiring supplemental oxygen, invasive mechanical ventilation, or ECMO. The EUA was based primarily on a phase III randomized double-blind trial, the ACTT-2, in which baricitinib improved the time to recovery when given in combination with remdesivir relative to remdesivir alone, in patients who required supplemental oxygen but not invasive mechanical ventilation. The FDA review also included data for baricitinib from the approved indication of rheumatoid arthritis and from populations studied for other indications, including pediatric patients.
Pharmacological Properties
Baricitinib is an orally administered, selective inhibitor of JAK1 and JAK2 that is approved for the treatment of rheumatoid arthritis. As a JAK–STAT signaling inhibitor, baricitinib inhibits the intracellular signaling pathway of cytokines known to be elevated in severe COVID-19, including IL-2, IL-6, IL-10, interferon-gamma (IFN-γ), and granulocyte-macrophage colony-stimulating factor. In addition, through the use of artificial intelligence–inspired algorithms, it was identified as a potential therapeutic because of its high affinity (half-maximal inhibitory concentration [IC 50 ] of 34 nM) against AP2-associated protein kinase 1 (AAK1), which may lead to interrupted SARS-CoV-2 cellular entry and intracellular assembly of viral particles , ( Fig. 16.4 ). The human pharmacokinetic (PK) profile of baricitinib is characterized by dose-proportional increases in systemic exposure and an elimination half-life of 12 hours. After once-daily administration, the unbound peak concentration (103 nM) at a 10-mg daily dose was projected to exceed the IC 50 for AAK1, further supporting evaluation in patients with COVID-19.

Dosing and Indication
Baricitinib is available as oral tablets. In the EUA for baricitinib in combination with remdesivir, the recommended dosage in adults with an estimated glomerular filtration rate (eGFR) ≥60 mL/min/1.73 m 2 is 4 mg once daily for 14 days of total treatment or until hospital discharge, whichever is first. The authorization to use in pediatric patients comes from data in other indications, with a recommended dose of 4 mg once daily for patients ≥9 years, and 2 mg once daily for patients aged 2 through <9 years, for 14 days or until hospital discharge. For both adults and pediatric patients, dosage adjustments are required for those with renal or hepatic impairment; the drug is not recommended for patients who are on dialysis, have end-stage renal disease, or have experienced acute kidney injury.
Clinical Efficacy
The EUA was based on data from a randomized double-blind, placebo-controlled trial (ACTT-2) in which patients received either remdesivir and baricitinib (n = 515) or remdesivir and placebo (n = 518). Randomization was stratified to trial site and disease severity at the time of enrollment. Remdesivir was administered by intravenous infusion as a 200-mg loading dose on day 1, then 100 mg daily from day 2 to day 10, or until hospital discharge or death. Baricitinib was administered as a 4-mg dose once daily for 14 days or until hospital discharge. Patients with an eGFR less than 60 mL per minute received baricitinib 2 mg once daily. Patients had to have laboratory-confirmed SARS-CoV-2 infection and at least one of the following to be enrolled in the trial: radiographic infiltrates by imaging, SpO 2 94% or less on room air, a requirement for supplemental oxygen, or a requirement for mechanical ventilation or ECMO. Mean age was 55 years (with 30% of patients aged 65 or older); 63% of patients were male, 51% were Hispanic or Latino, 48% were White, 15% were Black or African American, and 10% were Asian; 14% did not require supplemental oxygen, 55% required supplemental oxygen, 21% required noninvasive ventilation or high-flow oxygen, and 11% required invasive mechanical ventilation or ECMO.
The primary endpoint was median time to recovery defined as discharged from hospital or hospitalized but not requiring supplemental oxygen or ongoing medical care. For the overall population, the median time to recovery was 7 days for baricitinib plus remdesivir versus 8 days for placebo plus remdesivir (RR for recovery, 1.16; 95% CI, 1.01–1.32; P = .03) (see Table 16.1 ). In secondary analyses, by day 29, fewer patients died or progressed to noninvasive ventilation/high-flow oxygen or invasive mechanical ventilation with baricitinib plus remdesivir (22.5%) compared with remdesivir alone (28.4%) (RR, 0.77; 95% CI, 0.60–0.98). The Kaplan–Meier estimates of mortality at day 28 numerically favored the combination, with 5.1% (95% CI, 3.5–7.6) in the combination arm compared with 7.8% (95% CI, 5.7–10.6) with remdesivir alone, but the difference was not statistically significant (HR for death 0.65; 95% CI, 0.39–1.09). The upper bound of the confidence limit of the hazard ratio suggests that the combination was not likely to have an unacceptable increase in mortality. Additionally, the incidence of new use of oxygen was lower in the combination group than in the control group (22.9% vs. 40.3%; difference, −17.4 percentage points; 95% CI, −31.6 to −2.1), as was the incidence of new use of mechanical ventilation or ECMO (10.0% vs. 15.2%; difference, −5.2 percentage points; 95% CI, −9.5 to −0.9).
Clinical Safety
Although higher than the FDA-approved dose of 2 mg for rheumatoid arthritis, the dosing period for baricitinib 4 mg once daily for the treatment of COVID-19 was limited to 14 days and substantial safety information from indications other than COVID-19 was available at the 4-mg dose to inform the safety profile. Safety data for the combination was available for 507 patients hospitalized with COVID-19. In the ACTT-2 trial, fewer patients experienced serious adverse events in the combination arm (15%) compared with remdesivir alone (20%), and a similar pattern was seen with treatment-emergent adverse events (41.3% vs. 47.5%). The rate of discontinuations because of adverse events was also lower in the combination arm (6.7%) compared with remdesivir alone (11.6%). On the other hand, an increase in thrombotic events was observed in the combination arm (4%) versus 3%, with 5 patients experiencing serious pulmonary embolism compared with 1 patient with remdesivir alone. This is consistent with previous observations in patients with rheumatoid arthritis. Common nonserious adverse events were decreases in GFR and hypertension. There was no increase in the overall infections observed after 14 days of combination treatment compared with placebo. Overall, the safety profile observed in patients with COVID-19 in ACTT-2 was consistent with the established safety profile for baricitinib.
There were no clinical data for baricitinib in pediatric patients with COVID-19. Instead, the dosing recommendations were derived from these available sources and insights: (1) PK data in pediatric patients with juvenile idiopathic arthritis, atopic dermatitis, and in diseases referred to as type I interferonopathies; (2) data from adults with COVID-19; (3) considerations that the disease in adults and pediatric patients is sufficiently similar once patients progress to require supplemental oxygen, invasive mechanical ventilation, or ECMO; and (4) that there are no known COVID-19–specific pathophysiological differences (pediatric vs. adult), which can significantly affect the PK profile of baricitinib.
Neutralizing Antibody Combination: Casirivimab and Imdevimab
On November 21, 2020 the FDA issued an EUA for (REGEN-COV) casirivimab and imdevimab (administered together) for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) at high risk for progressing to severe COVID-19 and/or hospitalization. The basis of approval stemmed from a randomized double-blind, placebo-controlled clinical trial in nonhospitalized adults with mild to moderate COVID-19 symptoms, which showed that the combination reduced COVID-19–related hospitalization or emergency department (ED) visits in patients at high risk for disease progression compared with placebo.
Of note, the product is not authorized for patients hospitalized for COVID-19 or who require oxygen therapy as a result of COVID-19 because a beneficial effect of casirivimab and imdevimab treatment has not been shown in patients hospitalized for COVID-19. Moreover, mAbs such as casirivimab and imdevimab may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high-flow oxygen or mechanical ventilation.
Pharmacological Properties
Casirivimab and imdevimab (immunoglobulin G1λ [IgG1λ]) are recombinant human IgG1 mAbs, unmodified in the Fc regions. The mAbs are covalent heterotetramers consisting of two heavy and two light chains, produced by recombinant DNA technology in Chinese hamster ovary cell suspension cultures. They bind to nonoverlapping epitopes of the spike protein RBD of SARS-CoV-2 with an IC 50 of 81.8 pM , ( Fig. 16.5 ).

In a cell-based assay (Vero E6 cells), the combination neutralized SARS-CoV-2 with EC 50 of 31.0 pM (0.005 μg/mL). The combination was also shown to possibly mediate antibody-dependent cell-mediated cytotoxicity with natural killer cells and antibody-dependent cellular phagocytosis with macrophages. The human PK profiles of casirivimab and imdevimab are characterized by dose-proportional exposures (between 600- and 4000-mg) and mean estimated half-lives of 32 and 27 days, respectively.
Dosing and Indication
The authorized dosage is 1200 mg of casirivimab and 1200 mg of imdevimab administered together as a single intravenous infusion as soon as possible after a positive viral test for SARS-CoV-2, and within 10 days of symptom onset. The drugs are available in a co-formulated vial (in a 1:1 ratio) and also as individual vials. The combination can be subcutaneously administered in the event intravenous infusion is not feasible and would delay treatment. The optimal dosing regimen for treatment of COVID-19 has not yet been established; the authorized dosing regimen may be updated with additional data from clinical trials.
As noted earlier, the EUA is for mild to moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) who are at high risk for progressing to severe COVID-19 and/or hospitalization. Conditions that may place patients at higher risk for progression to severe COVID-19 include older age, obesity, diabetes, hypertension, chronic kidney disease, and others. The efficacy and safety of casirivimab with imdevimab have not been evaluated in hospitalized patients. Therefore the combination is not authorized for patients in these settings: (1) patients who are hospitalized, (2) patients who require oxygen therapy because of COVID-19, and (3) patients requiring an increase in baseline oxygen flow rate while on chronic oxygen therapy for non–COVID-19 conditions.
Clinical Efficacy
Mild to Moderate COVID-19 (R10933-10987-COV-2067)
The EUA was based on data from a phase I/II/III trial, COV-2067. In the phase III trial, adults with at least one risk factor for severe COVID-19 received 600 mg each of casirivimab and imdevimab (n = 838), 1200 mg of each mAb (n = 1529), 4000 mg of each mAb (n = 700), or placebo (n = 1500) in a randomized design. Based on phase I/II efficacy results, the phase III portion of the protocol was amended to compare a 1200 mg dose of each mAb versus placebo, and 600 mg dose of each mAb versus placebo.
At baseline, the median age was 50 years (with 13% of subjects ages 65 years or older), 52% of the subjects were females, 84% were White, 36% were Hispanic or Latino, and 5% were Black or African American. Among patients with baseline symptom data, 42% had severe symptoms, 42% had moderate symptoms, 15% had mild symptoms, and 2% reported no symptoms. The median duration of symptoms was 3 days; mean viral load was 6.2 log10 copies per milliliter at baseline. Demographics and disease characteristics at baseline were well balanced across the treatment groups.
The primary endpoint was the proportion of subjects with one or more COVID-19–related hospitalizations or all-cause death through day 29. The modified full analysis set was subjects with a positive reverse transcription polymerase chain reaction (RT-PCR) result and with at least one risk factor for severe COVID-19. As shown in Table 16.1 , COVID-19–related hospitalization or death occurred in 7 (1.0%) patients in the casirivimab and imdevimab 600-mg group compared with 24 (3%) in the placebo group, corresponding to a 70% reduction ( P = .0024). Similar results were observed for the higher dose, thus supporting the EUA of the 600-mg dose for both mAbs.
Clinical Safety
The safety of 600 mg casirivimab plus 600 mg imdevimab is based on an analysis from ambulatory (nonhospitalized) subjects with COVID-19 from the COV-2067 trial. In the phase III portion of the trial, treatment-emergent adverse events were observed in 59 (7%) patients in the 600-mg dose group of each mAb (n = 827) compared with 189 (10%) in the placebo group (n = 1843). Fewer patients reported serious adverse events in the 600-mg dose group (1%–2%) compared with placebo (4%). Infusion-related reactions of grade 2 or higher severity were observed in 2 subjects in the 600-mg dose group compared with none in the placebo group.
Key warnings and precautions include serious hypersensitivity reactions such as anaphylaxis and potential for severe infusion-related reactions, which may be mitigated with slowing or stopping the infusion. Additionally, clinical worsening of COVID-19 after administration of REGEN-COV has been reported, although it is not known if these events were related to drug or disease progression. Thus the combination is not authorized for use in patients hospitalized for COVID-19 who require oxygen therapy for COVID-19 or who require an increase in baseline oxygen flow rate because of COVID-19 when receiving chronic oxygen therapy for comorbidity not related to COVID-19.
Neutralizing Antibody Combination: Bamlanivimab and Etesevimab
On February 9, 2021 the FDA issued an EUA for emergency use of bamlanivimab and etesevimab administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The EUA was based on the phase II/III BLAZE-1 trial, a randomized double-blind, placebo-controlled clinical trial.
Pharmacological Properties
Bamlanivimab and etesevimab are neutralizing IgG1 mAbs that bind to distinct but overlapping epitopes within the RBD of the spike protein of SARS-CoV-2. The antibodies were derived from two patients who recovered from COVID-19 in North America and China. ,
SARS-CoV-2 neutralizing antibody discovery efforts have focused on targeting the multidomain surface spike protein, a trimeric class I fusion protein that mediates viral entry. The viral entry depends on the interaction between the RBD and the ACE2 cellular receptor. Antibodies that bind the RBD and interfere with ACE2 binding can have potent neutralizing activity. Bamlanivimab is a recombinant neutralizing human IgG1κ mAb to the spike protein of SARS-CoV-2, unmodified in the Fc region. Bamlanivimab binds the spike protein with a dissociation constant (KD) = 0.071 nM and blocks spike protein attachment to the human ACE2 receptor with an IC 50 value of 0.17 nM (0.025 µg/mL). Etesevimab is a recombinant neutralizing human IgG1κ mAb to the spike protein of SARS-CoV-2, with amino acid substitutions in the Fc region (L234A, L235A) to reduce effector function. Etesevimab binds the spike protein with a dissociation constant KD = 6.45 nM and blocks spike protein attachment to the human ACE2 receptor with an IC 50 value of 0.32 nM (0.046 µg/mL (see Fig. 16.5 ). The human PK profiles of bamlanivimab and etesevimab are dose proportional between 700- and 7000-mg doses, with half-lives of 18 days and 25 days, respectively.
Dosing and Indication
The authorized dosages for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) is bamlanivimab 700 mg with etesevimab 1400 mg. The recommendation is to administer the antibodies as soon as possible after a positive viral test for SARS-CoV-2 and within 10 days of symptom onset. Under this EUA, bamlanivimab and etesevimab are diluted and administered together as a single intravenous infusion. Different dilutions are needed for patients weighing 50 kg or more versus patients weighing less than 50 kg.
Clinical Efficacy
Mild to Moderate COVID-19 (BLAZE-1): Phase II Portion
The design of the phase II portion of BLAZE-1 included subjects receiving a single infusion of bamlanivimab 2800 mg and etesevimab 2800 mg (N = 112), bamlanivimab alone (at doses of 700 mg [N = 101], 2800 mg [N = 107], or 7000 mg [N = 101]) or placebo (N = 156). The primary endpoint was the change in viral load from baseline to day 11. For the combination treatment, the differences in the change in log viral load at day 11 compared with placebo were 0.09 (95% CI, –0.35 to 0.52; P = .69) for 700 mg, –0.27 (95% CI, –0.71 to 0.16; P = .21) for 2800 mg, 0.31 (95% CI, –0.13 to 0.76; P = .16) for 7000 mg, and –0.57 (95% CI, –1.00 to –0.14; P = .01). The proportion of patients with COVID-19–related hospitalizations or ED visits was 5.8% (9 events) for placebo, 1.0% (1 event) for 700 mg, 1.9% (2 events) for 2800 mg, 2.0% (2 events) for 7000 mg, and 0.9% (1 event) for combination treatment, supporting the activity of these agents.
Mild to Moderate COVID-19 (BLAZE-1): Phase III Portion
In the phase III portion, subjects received a single intravenous infusion of bamlanivimab 700 mg and etesevimab 1400 mg (N = 511) or placebo (N = 258). The median age was 56 years (30% aged 65 years or older), 53% were females, 87% were White, 27% were Hispanic, and 8% were African American. Majority of the subjects had mild COVID-19 (76%) and the rest (24%) had moderate disease. The primary endpoint, proportion of subjects with COVID-19–related hospitalization (defined as ≥24 hours of acute care) or death by any cause by day 29, occurred in 6% of subjects in the placebo arm (15 subjects) compared with 0.8% (4 subjects) in the experimental arm, corresponding to an 87% reduction ( P < .0001) (see Table 16.1 ). Counting just the 29-day mortality alone, there were 4 deaths in the placebo arm and no deaths in subjects treated with bamlanivimab 700 mg and etesevimab 1400 mg together ( P = .01).
Clinical Safety
The safety of bamlanivimab administered with etesevimab is primarily based on exposure of approximately 1400 ambulatory (nonhospitalized) subjects who received doses of bamlanivimab and etesevimab together, at the recommended dose of (bamlanivimab 700 mg, etesevimab 1400 mg) or higher. Safety data for the 700-mg bamlanivimab and 1400-mg etesevimab combination has not been reported at the time of writing this chapter. In the phase III trial, adverse events occurred in 13% of subjects who received 2800 mg of bamlanivimab plus 2800 mg of etesevimab and in 12% of placebo-treated subjects. The combination is generally well tolerated. The most common treatment-emergent adverse events observed with bamlanivimab plus etesevimab have been nausea, dizziness, and pruritus, and none occurred in more than 1% of participants. Across ongoing, blinded clinical trials, a case of anaphylaxis and other cases of infusion-related reactions (n = 16, 1.1%) have been reported with the combination, which resolved upon stopping infusion and treatment (once with epinephrine).
The key warnings and precautions to take with the combination are related to hypersensitivity reactions. Although clinical worsening of COVID-19 symptoms has been observed with the combination, it is not known whether it was related to the combination or disease progression. Thus the combination is not authorized for use in patients hospitalized for COVID-19, who require oxygen therapy for COVID-19, or who require an increase in baseline oxygen flow rate because of COVID-19 when receiving chronic oxygen therapy for comorbidity not related to COVID-19.
Repurposed Drugs and New Molecular Entities in Clinical Development
In addition to medicines authorized under an EUA program or that have full approval in at least one country (remdesivir), numerous organizations are involved in developing new molecular entities or in repositioning or repurposing medicines previously approved for other indications to address processes that drive the pathogenesis of COVID-19. These processes include the replication of SARS-CoV-2 early on during the disease process and the dysregulated immune and inflammatory response to SARS-CoV-2 that leads to tissue and organ damage ( Fig. 16.6 ).

One of the most successful repurposing efforts has been the use of corticosteroid therapy, including primarily dexamethasone (and to a lesser extent, methylprednisolone), to manage the severe complications associated with the immune and inflammatory response phase of COVID-19. Therefore this section begins with an overview of dexamethasone similar to that provided for drugs with EUA.
Dexamethasone
On September 2, 2020, the WHO issued a living guidance entitled “Corticosteroids for COVID-19.” The interim guideline suggests two recommendations regarding the use of corticosteroids according to COVID-19 disease severity, based on a prospective meta-analysis of randomized trials for corticosteroid therapy for COVID-19. For patients with severe and critical COVID-19, the WHO recommends the use of systemic (i.e., intravenous or oral) corticosteroids. For patients with nonsevere COVID-19, the WHO suggests not to use corticosteroids.
Research findings on dexamethasone, a corticosteroid, from the United Kingdom’s national Randomised Evaluation of COVID-19 Therapy (RECOVERY) trial was the main evidence initiating and supporting the WHO recommendations. The use of dexamethasone resulted in lower 28-day mortality among patients with COVID-19 who required invasive mechanical ventilation or oxygen therapy, but not among patients who did not receive respiratory support at randomization.
Pharmacological Properties
The general rationale for the use of dexamethasone for patients with severe COVID-19 is related to the pathogenesis of the disease. In the later disease phase, patients with COVID-19 suffer from organ tissue damage that is mainly driven by a dysregulated immune and inflammatory response (i.e., a hyperinflammatory state or cytokine storm) as a result of SARS-CoV-2 infection. , The use of corticosteroids for antiinflammatory and immunosuppressant effects may reduce organ tissue damage that can lead to long-term health problems (e.g., heart failure, long-term breathing problems, strokes, and even mortality. Dexamethasone has potent glucocorticoid effects: increasing the production of antiinflammatory compounds, decreasing the production of proinflammatory compounds, and increasing apoptosis in inflammatory cells, by suppressing the migration of neutrophils and decreasing lymphocyte proliferation , ( Fig. 16.7 ).

Dexamethasone (9α-fluoro-16α-methylprednisolone) was synthesized in 1957 because of the need for a steroid with a longer duration of action. The biological half-life of dexamethasone is 36 to 72 hours. Oral doses of dexamethasone in the 1.5- to 6-mg dose range have a half-life of approximately 7 hours; a 20-mg oral tablet has a half-life of 4 hours, and a 4-mg intravenous dose has a half-life of 9 hours. , The absolute bioavailability of dexamethasone is 81% (95% CI, 54–121) in healthy subjects.
Dosing and Indication
Dexamethasone is available in various formulations, including tablets with a range of 0.5 to 6 mg, an oral solution with a range of 0.5 mg/5 mL to 1 mg/mL, and injectable suspension with a range of 4 mg/mL to 20 mg/5 mL. The glucocorticoid effect of 1 mg of dexamethasone is equivalent to that of 8 mg of prednisolone or 25 mg of hydrocortisone. A general dosing recommendation for the treatment of inflammation for adults whose age is 18 years or older is to start with a dose of 0.75 mg daily and to increase to 9 mg daily, with doses divided two or four times daily. , For children ages 17 years or younger, 0.02 to 0.3 mg per kilogram of body weight per day is recommended, with dosing divided three or four times daily. As with other drugs, a lower dose or less frequent dosing schedule would be needed for seniors, with consideration of their kidney and liver function. In addition, for a life-threatening condition, 10 mg of intravenous dexamethasone, followed by 4 mg of intramuscular administration given every 6 hours is recommended, with tapering over 7 days to discontinue the therapy. Although the use of dexamethasone is recommended for patients with severe COVID-19, it remains unclear what the most appropriate dose would be in its treatment.
Clinical Efficacy
The RECOVERY trial is a multicenter, randomized, open-label study assessing 28-day mortality as the primary outcome among 6425 hospitalized patients with COVID-19. The use of dexamethasone (6 mg once daily for up to 10 days) reduced the 28-day mortality most significantly for patients who required invasive mechanical ventilation at randomization (28-day mortality: 29.3% in dexamethasone arm vs. 41.4% in control arm; rate ratio, 0.64; 95% CI, 0.51–0.81). Some extent of the survival benefit was also observed among patients who received oxygen therapy but did not require invasive mechanical ventilation (28-day mortality: 23.3% in dexamethasone arm vs. 26.2% in control arm; rate ratio, 0.82; 95% CI, 0.72–0.94). However, the use of dexamethasone did not reduce the 28-day mortality rate among patients who did not require any respiratory support at randomization (28-day mortality: 17.8% in dexamethasone arm vs. 14.0% in control arm; RR, 1.19; 95% CI, 0.92–1.55).
Other corticosteroids have been investigated for COVID-19 and compared with the effectiveness of dexamethasone. Recently, a finding from a prospective triple-blinded randomized controlled trial was published, which demonstrated methylprednisolone (2 mg/kg/day) showed significantly better clinical outcomes compared with dexamethasone (6 mg/day) among a total of 86 patients who were hospitalized for COVID-19. The study used a 9-point WHO ordinal scale from 0 (uninfected) to 8 (death), with the lower the score, the better. The methylprednisolone group showed significantly lower values at day 5 (4.02 in methylprednisolone arm vs. 5.21 in the dexamethasone arm, P = .002) and day 10 (2.90 in methylprednisolone arm vs. 4.71 in dexamethasone arm, P = .001) of admission. The mean length of hospital stay (7.43 ± 3.64 days in the methylprednisolone arm vs. 10.52 ± 5.47 days in the dexamethasone arm, P = .015) and the need of a ventilator (18.2% in the methylprednisolone arm vs. 38.1% in the dexamethasone arm, P = .040) also supported the conclusion.
Clinical Safety
Adverse Events
Dexamethasone is generally tolerated. , , Its short-term use is not associated with any serious adverse drug effects or allergic reactions. The most common reported adverse drug effect is insomnia (sleep disorder), along with agitation, depression, acne, increased appetite, indigestion, nausea, and vomiting. However, its long-term use (i.e., >2 weeks) may be associated with psychological problems (e.g., irritation, mood swings, and memory issues), vision problems (e.g., glaucoma and cataract), bone and joint pain (i.e., increasing risk for osteoporosis), signs of infections (e.g., fever and sore throat), or symptoms of intestinal bleeding (e.g., abdominal pain and dark stools). In particular, dexamethasone is not recommended for patients who have a history of infectious diseases, such as tuberculosis, fungal infections, and parasite infections, because it may hide and worsen the harmful effects of certain infections.
Drug–Drug Interactions
Antibiotics (e.g., erythromycin) and antifungal drugs (e.g., ketoconazole, itraconazole, posaconazole, and voriconazole) can increase the plasma concentration of dexamethasone, leading to higher risk for adverse drug reactions of dexamethasone. In contrast, the use of dexamethasone can decrease the concentration of anticoagulants (e.g., apixaban and rivaroxaban), leading to higher risk for a blood clot or stroke. Dexamethasone can also decrease the efficacy of human immunodeficiency (HIV) drugs (e.g., protease inhibitors such as ritonavir and nonnucleoside reverse transcriptase inhibitors such as etravirine) and tuberculosis drugs (e.g., isoniazid).
Special Populations
Dexamethasone may delay a child’s growth. For pregnant or breastfeeding women, adverse drug reactions of dexamethasone have not been studied enough, but animal research showed a risk for side effects to the fetus.
Other Repurposed Medicines With Randomized Controlled Trial Data
A useful framework was proposed by Khani et al. for COVID-19 therapeutics by adapting the classification system recommended by the American College of Cardiology/American Heart Association based on strength of recommendation (Class I = strong benefit, Class IIa = moderate benefit, Class IIb = weak benefit, and Class III = no benefit or harmful) and quality of evidence (A = high-quality RCT, B-R = moderate-quality RCT, B-NR = moderate-quality non-RCT, C-LD = limited data, and C-EO = expert opinion). Based on this classification and considering that a number of investigational therapeutics besides those that have received EUA or approval have been evaluated in an RCT, a summary of the outcomes is provided in Table 16.2, modified from the review by Khani et al. Agents with non-RCT data have not been considered, given the high potential to yield biased or misleading results.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


