Abstract
Management of breast cancer has always been informed by our understanding of the biology of this disease. As we moved from descriptive models to biologically grounded models, we began designing treatments that take advantage of the weaknesses of this cancer. The hallmark of this cancer is its heterogeneity between patients and within the same patients at different stages of the disease. Traditional classifiers and modern genomic tools allow the classification of breast cancer into subtypes that instruct its treatment that has become extremely different from one type to the other and from one patient to the other. Surgery has become less invasive and radiation therapy less toxic. Chemotherapy, hormonal therapy, targeted therapy and immunotherapy have resulted in dramatic improvement of outcomes for many patients and has led to decreased mortality in the face of increasing incidence of the disease. This chapter addresses our new understanding of breast cancer and highlights the major advances in its treatment.
Keywords
Breast cancer, intrinsic subtypes, tumor heterogeneity, centrifugal model, systemic model, progressive model, indolent model, tumor dormancy, disseminated tumor cells, surgical treatment, radiation therapy, chemotherapy, endocrine therapy, targeted therapy, immunotherapy
No cancer has seen so many dramatic changes in its care and so much passion in the discussions surrounding these changes as breast cancer. In one century, we moved from the aggressive widely mutilating Halstedian surgery to the Z-11 era where up to 25% of patients are left with lymph node metastasis in the axilla without any added recurrence or mortality risk, provided that radiation is given to them. It is clear that we have come a long way in our practice but also in our understanding of this cancer. This magnificent progress occurred most of the time incrementally, as a result of slow scientific discoveries in basic and clinical sciences, but occasionally in bursts that resulted from the advent of scientific breakthroughs, new ideas, and new paradigms to explain cancer in general and the natural history of breast cancer in particular.
Epidemiology of Breast Cancer
Globally, breast cancer is the most common cancer in women and the second most common cancer in the world. More than 1.67 million new breast cancer cases were diagnosed in 2012 (25% of all cancers). Incidence rates range from 27 per 100,000 in developing areas (Middle Africa and East Asia) to 92 in more developed areas (North America). Several epidemiologic studies have linked these different rates to reproductive (parity) and breastfeeding differences in addition to lower screening rates and incomplete reporting. If women in developed countries had the average number of births and lifetime duration of breastfeeding that women in developing countries have had until recently, the cumulative incidence of breast cancer would be reduced by more than half, from 6.3 to 2.7 per 100 women by age 70. The protective effect of parity was restricted to estrogen receptor (ER)-positive/progesterone receptor (PR)-positive breast cancer with each birth reducing the risk of ER-positive PR-positive cancer by 11% (relative risk per birth = 0.89, 95% confidence interval [CI] 0.84–0.94).
Even though incidence rates are much lower in developing countries, mortality rates are practically equal across the world due to less favorable health conditions and limited access to care in the developing countries (324,000 deaths, 14.3% of total, in developing countries vs. 198,000 deaths, 15.4% of total, in developed countries). Worldwide, the incidence of breast cancer has been going up in most regions but at faster rates in developed than developing countries. Trends in mortality from breast cancer, however, show steady decrease in most developed countries and stagnation or slight increase in developing countries.
Incidence rates and mortality from breast cancer in the United States have gone through different phases of increase and decline starting in 1943. The increase was slow between 1943 and 1979 by 1% a year, then more rapid from 1980 to 1999 at 4%. It plateaued after 2003 after a gradual (2000–2002) then sharp decline between 2002 and 2003. The rapid increase after 1980 is likely related to increased use of hormone replacement therapy (HRT) and the wide introduction of screening mammography, and the drop after 2002–2003 was explained by a 38% drop in the use of HRT after the publication of the Women’s Health Initiative study. This increase in incidence was practically exclusively related to in situ and localized ER-positive/PR-positive cancers. In the Connecticut experience, which mirrors the whole US experience, in situ and localized cancers increased by 1023% and 86%, respectively. This increase was not associated with a proportional decrease of regional and distant disease (−15% and −20%, respectively).
Between 1975 and 1990, mortality rates continued rising by 0.4% a year, then started declining starting in 1990 by 2.2 % annually until now. Until 1970, mortality rates in the United States were similar for blacks and whites and have been declining since the late 1980s, among whites first, then during the 1990s among blacks.
Breast Cancer Risk Factors
Breast cancer risk factors include age, reproductive factors, breastfeeding, oral contraceptives, HRT, diet, obesity, smoking, alcohol, and heredity ( Table 24.1 ).
| Relative Risk | Reproductive Factors | Cancer Type |
|---|---|---|
| 1.4 | Age >40 | Luminal A |
| 1.5 | Age >40 | HER2 |
| 1.48 | Nulliparous | Luminal type |
| >4 | Postmenopausal breast density | >4 |
| 0.5 | Full-term pregnancy before age 20 | 0.5 |
| Increased | Younger age at menarche (<12 y) | |
| 0.03 for every additional year | Older age of menopause (>55 y) | |
| 0.74 | Breastfeeding | Less luminal |
| Oral contraceptives: | ||
| 1.5 | Lifetime use ≥15 y | |
| 1.6 | Current user ≥5 y | |
| 3.5 | Current use for ≥5 y among ages 20–39 | Especially TNBC |
| 1.9 | HRT (>5 y) especially E-P > E | Luminal A |
| 2.2 | Hormone replacement therapy (>5 y) especially E-P > E | HER2 |
| Overweight (BMI 25–30); obesity (BMI >30) | ||
| 1.89 | Premenopausal | TNBC |
| 0.63 | Premenopausal | Luminal |
| Increased | Postmenopausal | Luminal |
| Decreased | Oophorectomy at age <40 y is protective | |
| Increased | Diet (high fat) | |
| Increased | Smoking | |
| 1.2 | Alcohol ≥2 drinks a day | |
| Increased | Heredity | |
| Hereditary breast cancer syndromes | ||
| 6 | BRCA1 | TNBC |
| 5 | BRCA2 | Same as sporadic |
| Increased | Familial syndromes | Same as sporadic |
A rapid review of these risk factors would classify them into three categories: (1) increased and prolonged estrogenic stimulation (early menarche, late menopause, nulliparity, late first full-term pregnancy, oral contraceptives, HRT, obesity in postmenopausal women) increases the risk of breast cancer. Decreased estrogenic stimulation (multiparity, breastfeeding, early menopause, obesity in premenopausal women) decreases the risk of breast cancer. (2) Increased exposure to carcinogens (tobacco, alcohol, others) leads to increased risk of breast cancer. (3) Decreased ability to repair genetic damage or to eliminate cells that harbor them (mutation of BRCA1 and BRCA2, PTEN, p53, and others) leads to higher incidence of breast cancer.
The Natural History of Breast Cancer
Traditional Models for Breast Cancer Natural History
In the beginning of the 20th century, the prevailing model for the natural history of breast cancer at the time of Halsted was the centrifugal model. This model stipulates that cancer starts in the breast and moves slowly but surely in an orderly, defined manner to invade the breast tissue and surrounding structures then to the lymph nodes, and, if it is not radically removed, then it will spread out from the locoregional basin to distant organs, always by contiguous spread. The involvement of the lymph nodes was conceived to happen by direct extension, and the lymph nodes were viewed as a true barrier to the progression of the cancer. Hematogenous spread was given little importance, and tumors were considered independent of the host. This gradual stepwise progression and understanding of the biology of cancer borrowed from infectious diseases dictated an approach to the management of breast cancer that was based on “en bloc” wide surgical resection of the breast, muscles, fat, and lymph nodes to remove all the cancer without “contaminating” healthy tissues on the side of the affected breast. This is the radical mastectomy promoted by Halsted and Meyer at the turn of the 20th century. The success of breast cancer surgery was determined by the extent of tissue removed. Women undergoing this surgery were left with horrendous morbidities such as limited arm mobility, severe lymphedema, and major deformities of body anatomy and image. Despite increasingly aggressive surgeries, women kept progressing to metastatic stages and dying of breast cancer. In the 1950s and 1960s, clinical and basic research led many investigators to question this model.
Debunking the centrifugal model occurred after demonstrating that many of its premises were inaccurate. The first to be discredited was the idea that lymph nodes are effective barriers that stop the spread of tumor cells. Cancer cells injected in the afferent lymphatic of the leg lymph nodes of an animal were collected in the efferent lymph draining from the node. This crude experiment evaluated the question from a mechanical standpoint and did not take into consideration the possibility of interaction between the immune system and the cancer cells that may need time and other conditions to occur. Nevertheless, it was adequate to start shattering the model by causing the first crack in it. The role of the blood as a “highway” for cancer cell dissemination was ascertained in other experiments and led to the conclusion that recurrences are not the result of local treatment failure but of reactivation of dormant disseminated tumor cells that develop from the interaction between the tumor and the host. An “alternative hypothesis” was proposed to account for the aspects that the Halstedian model was unable to explain and to take into consideration these findings. This is the “systemic hypothesis” that suggested that breast cancer is a systemic disease from its inception. The involvement of the lymph nodes is nothing else but an indication that the cancer cells can metastasize and colonize distant organs rather than the instigator of disseminated disease ( Table 24.2 ).
| TWO DIVERGENT HYPOTHESES OF TUMOR BIOLOGY | |
|---|---|
| Halstedian | Alternative |
| Tumors spread in an orderly defined manner based on mechanical considerations. | There is no orderly pattern of tumor cell dissemination. |
| Tumor cells traverse lymphatics to lymph nodes by direct extension supporting en bloc dissection. | Tumor cells traverse lymphatics by embolization challenging the merit of en bloc dissection. |
| The positive lymph node is an indicator of tumor spread and is the instigator of distant disease. | The positive lymph node is an indicator of a host-tumor relationship, which permits development of metastases rather the instigator of distant disease. |
| Regional lymph nodes are barriers to the passage of tumor cells. | Regional lymph nodes are ineffective as barriers to tumor cell spread. |
| RLNs are of anatomic importance. | RLNs are of biological importance. |
| The bloodstream is of little significance as a route of tumor dissemination. | The bloodstream is of considerable importance in tumor dissemination. |
| A tumor is autonomous of its host. | Complex host-tumor interrelationships affect every facet of the disease. |
| Operable breast cancer is a locoregional disease. | Operable breast cancer is a systemic disease. |
| The extent and nuances of operation are the dominant factors influencing patient outcome. | Variations in locoregional therapy are unlikely to substantially affect survival. |
The systemic model stipulates that the stage of the local disease at diagnosis and the extent of local therapy do not alter the natural history of the disease and are unlikely to change the incidence of metastatic recurrences. Several observations with longitudinal follow-up have suggested otherwise. The likelihood of distant metastasis was found to highly correlate with tumor size and the number of involved lymph nodes. On the other hand, the optimization of local surgical treatment by the addition of radiation therapy decreased both local and distant metastasis. The sharp dichotomy between the centrifugal model and the systemic model left a large number of tumors out of either model, which suggested to many investigators a different model, the spectrum hypothesis, to account for these discrepancies. The main premise of the spectrum hypothesis is the fundamental biological heterogeneity of breast cancer with variable progression speeds and possibilities to metastasize at different sizes. Some tumors are metastatic from their inception, and early local treatment does not affect the ultimate outcome (these tumors follow the systemic model); others may grow to large sizes, gaining the ability to spread to lymph nodes or distant organs only later during their course (these follow the centrifugal model). Early local treatment is likely to cure those patients. However, most tumors reach the metastagenicity stage at different points that vary depending on the specific biology of the tumor. Those tumors follow the progressive model and can be cured with local treatment only if detected early enough. Finally, some tumors never get there ( Fig. 24.1 ).
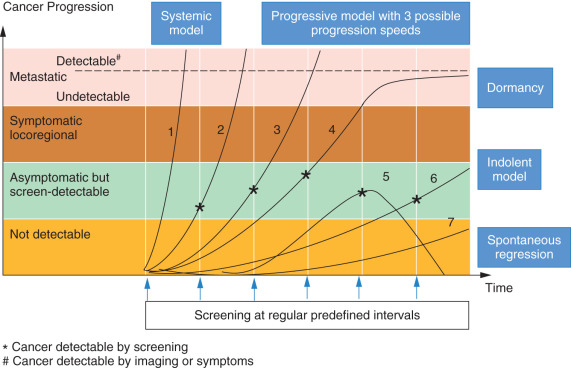
The relationship between tumor size and the probability of metastatic dissemination is linear with a median (V50) of 23.6 mL (diameter = 3.56 cm) and a 95% CI 0.14 to 4000 mL. This volume is smaller if the lymph nodes are involved and if tumor grade is high. The spectrum hypothesis suggests that metastatic dissemination is possible at any tumor size and it is influenced by growth rate. The inverse correlation with the number of lymph nodes involved is the result rather than the cause of the early and more vigorous dissemination that leads to lymph node seeding at small volumes. The variability of V50 is responsible for the different models presented in Table 24.3 .
| Group | No. Patients | V50 (mL) | Corresponding Diameter (cm) | Variation Interval (mL) | Median Delay Between Diagnosis and Detection of First Distant Metastasis (Months) |
|---|---|---|---|---|---|
| Overall | 2648 | 23.6 | 3.56 | 19.3–28.8 | |
| Histologic grade known (total) | 1596 | 41.0 | 4.27 | 30.5–54.8 | |
| 1 | 298 | 584.0 | 10.4 | 191–1765 | 65 |
| 2 | 766 | 29.5 | 3.83 | 19.5–44.7 | 44 |
| 3 | 532 | 23.0 | 3.53 | 14.6–35.0 | 21 |
| No. axillary lymph nodes invaded known (total) | 1722 | 32.8 | 3.97 | 24.5–43.8 | |
| 0 | 560 | 690.0 | 11.0 | 217–2180 | 69 |
| 1–3 | 657 | 30.3 | 3.87 | 19.0–48.4 | 43 |
| >3 | 505 | 7.2 | 2.40 | 4.0–13.1 | 30 |
This descriptive modeling does not explain the relationship between size and dissemination potential (why do tumor cells disseminate when the tumor reaches certain size?), nor does it tell us why for certain tumor size and growth rate only 50% of the tumors would disseminate not all of them. However, these models were crucial to help physicians design screening and treatment strategies to manage patients. Their validity (albeit partial) was confirmed by the results of modern screening and treatment protocols.
The Estrogen Paradox
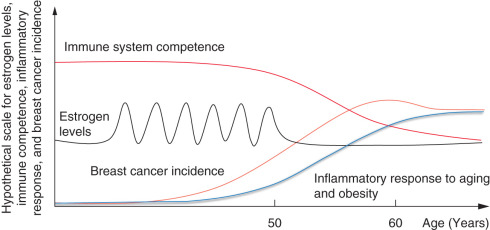
From the review of its risk factors, it is clear that breast cancer is an estrogen-driven cancer. However, breast cancer as a disease does not appear until estrogen secretion is depleted in a woman’s body. The peak incidence of breast cancer is age 62 years—10 years after the median age of menopause—and fewer than 5% of all breast cancers occur below age 50 when estrogen levels are the highest. Several hypotheses may be formulated to explain this paradox. Every menstrual cycle is an opportunity to induce proliferation in the mammary epithelial cell compartment with a possibility for genetic and epigenetic errors (gain of function mutations of oncogenes, loss of function of tumor suppressor genes, methylation of tumor suppressor genes) the probability of which gets higher with higher proliferation rate and higher number of proliferation cycles. The presence of a defect in the DNA repair system ( BRCA or p53 mutations) increases this probability. A classical autopsy series discovered small foci of subclinical breast cancers (mostly in situ) in 39% of women age 40 to 49 years who died from accidental death, whereas the incidence of clinical breast cancer in this age group is around 1%. Interestingly, many of these patients had multiple foci of preclinical cancers, and yet lifetime breast cancer incidence is not higher than 12% to 13% in our country. Why do these cancers remain silent and not progress to the open disease state?
To progress to clinically open disease, initiated tumor cells have to overcome multiple internal and systemic resistance mechanisms (apoptosis, lack of angiogenic switch, microenvironmental suppression or immune suppression of cancer cells). Indeed, DNA lesions induced by the estrogenic overdrive or carcinogens are necessary to induce malignant transformation but insufficient to induce progression to cancer disease. The progression of these lesions to become open cancers may require more DNA damage (that occurs over time from increased inflammatory response secondary to aging or obesity), initiation of angiogenesis (inflammatory cells may help initiate angiogenesis) and/or weakening of the immune system ( Fig. 24.2 ).
From Descriptive Models to Biologically Informed Carcinogenesis
Breast Cancer Initiation
Epidemiologic and experimental evidence lend a strong support to the role of estrogen in the initiation and progression of breast cancer. Cells in the normal mammary gland are subject to high turnover orchestrated by endocrine factors (estrogen, progesterone, prolactin, and others) in preparation for lactation during reproductive age. Every menstrual cycle is marked by significant proliferative changes of the ductal-lobular units; in the absence of pregnancy, massive cell death ensues. The process requires the recruitment of normal mammary stem cells that reside in the ducts. These cells are capable of asymmetric division in which one cell rejoins the pool of stem cells, and the other continues proliferating and differentiating to give rise to all the component of the mammary gland: the luminal and myoepithelial cells. Random or inherited genetic and epigenetic changes confer proliferative and/or survival advantages on certain mammary cells. These cells are believed either to belong to the stem cell compartment or to the progenitor cells after they have acquired stemness characteristics.
These incipient cancer cells face internal and external control mechanisms that, when successful, lead to the correction of the abnormalities, growth suppression of the cells, or their elimination altogether. The immune system plays a host-protective role in cancer control by detecting transformed cells and eliminating them, but it can, in certain circumstances, be tumor promoting.
Under the pressure of the immune system, transformed cells undergo many changes called immunoediting. There are three phases of immunoediting: elimination, equilibrium, and escape. Elimination is the process of eradication of the new cancer cells by a normal immune system and is supported by a wealth of experimental evidence in animals and humans. The innate and adaptive arms of the immune system recognize incipient cancer cells by the neoantigens (resulting from mutations or translocations) presented on their surface or by the distress signals that are expressed by transformed cells that have undergone chromosomal changes (aneuploidy or hyperploidy) and eliminate them. Equilibrium is reached when the immune system fails to eliminate the transformed cells but stop them from progressing further. It can be conceived as the dormancy phase of cancer development. This phase is mediated by equilibrium between cells and cytokines that promote elimination (interleukin [IL]-12, interferon [IFN]γ, tumor necrosis factor [TNF]α, CD4 Th1, CD8 + T cells, natural killer cells, γδT cells) and those that promote persistence of the nascent tumor (IL-23, IL-6, IL10, transforming growth factor-β, natural killer T cells, CD4 Th2, Foxp3 + T regulatory cells (Tregs), and myeloid-derived suppressor cells). Monocytes play an important role in this process. Under the influence of tumor microenvironment, they may differentiate into proinflammatory M1 or antiinflammatory M2 types, which play a role in the angiogenic switch (discussed in the next section).
Immune escape of cancer cells occurs by different mechanisms. In hormone receptor (HR)-positive breast cancer, the absence of strong tumor neoantigens and low expression of MHC1 allow the tumor to progress unnoticed by the immune system. Estrogen plays an immunosuppressive role in the tumor microenvironment that promotes tolerance of the weakly immunogenic cancer. In HER2 breast cancer, MHC1 presentation is inversely correlated with HER2 expression. Triple-negative breast cancer (TNBC) exhibits a spectrum of MHC1 presentation and strong tumor antigen expression but immune escape in this subtype is mostly related to the development of immunosuppressive tumor microenvironment by expressing immune checkpoints such as CTLA4 or PD-L1.
However, it is still unclear how the balance established during the equilibrium phase gets tilted toward tumor progression. The answer to this question is very likely multifactorial. Aging is associated with reduced production of new B and T lymphocytes in the bone marrow and the thymus, respectively and with decreased function of the mature ones. Systemic inflammation associated with aging and local proinflammatory microenvironment in the breast are incriminated in promoting cancerous progression of mammary stem cells that have been primed by losing tumor suppressor genes. Proinflammatory cytokines (TNFα and IL-6) were associated with overexpression of COX2 and the aromatase enzyme, which lead to increased local concentrations of estrogens. Estrogens induce the expansion of Tregs and the inhibition of antigen presenting cells. In addition to the gradual decline of the immune system, dietary, commensal microbiota, use of antibiotics, procreational, and hormonal factors, all play some role of variable importance in tilting the balance from equilibrium to escape.
The Vascular Phase of Tumor Development: The Angiogenic Switch
The prevascular stages of breast cancer precursor lesion, such as carcinoma in situ, can usually be seen only with microscopic examination. These tumor cells are also usually separated from host microvessels by a basement membrane. From experimental studies, we know that these prevascular lesions exist in a steady state of tumor cell proliferation balanced by cell death and may remain in this state for months to years. The onset of neovascularization, however, can be relatively sudden and is called the angiogenic switch. In most human breast cancers, angiogenesis is initiated after the emergence of the invasive malignancy, but it can also start at the preinvasive phase. It is understood as a shift in the net balance between positive regulators of angiogenesis (e.g., basic fibroblast growth factor [FGF], vascular endothelial growth factor [VEGF]) and negative regulators of angiogenesis (e.g., thrombospondin-1, 16-kD prolactin, IFN-α, IFN-β, platelet factor 4, angiostatin, endostatin, and others such as IL-12). Tumor-associated macrophages that were polarized to the M2 phenotype and other cells (fibroblasts and adipocytes) participate in generating proangiogenic factors that participate in the angiogenic switch.
Four mechanisms for angiogenic switch were identified. First, direct recruitment of blood supply by prevascular tumors. After the basement membrane is breached, invasive cells enter the stroma and induce neighboring endothelial cells to proliferate toward the source of proangiogenic factors following a concentration gradient. Multiple cell layers form a microcylinder (the radius of which is restricted by the oxygen diffusion limit) that surrounds every new capillary vessel. Second, recruitment of endothelial stem or progenitor cells. VEGF receptor-1–positive bone marrow cells are recruited from the blood to the invasive focus. However, the contribution of these cells in breast cancer angiogenesis is limited. Third, vessel cooption; cancer cells surround preexisting blood vessels. The last mechanism of angiogenic switch is vascular mimicry; the tumor cells dedifferentiate into an endothelial phenotype and make tubelike structures.
Angiogenesis is credited with allowing the tumor to grow from microscopic to macroscopic sizes and to facilitate the shedding of isolated cells or clusters of cells into the bloodstream, which leads to metastasis. New blood vessels provide the tumor with oxygen and the necessary nutrients and removal of waste products. Endothelial cells, even without blood flow, can provide the tumor with paracrine growth and survival signaling. The final effect of angiogenic switch is decreased apoptosis in the tumor by up to sevenfold while proliferation rates remain similar to the ones in the prevascular stage, which allows the rapid expansion of the tumor mass.
The inability of a tumor to turn the angiogenic switch on results in small microscopic tumors that remain dormant (angiogenic dormancy). Tumor dormancy is common in primary breast cancer and in metastatic disease and may extend to several years or decades. Chronic inflammation and the decline of the immune system in addition to the accumulation of genetic/epigenetic hits ultimately break the equilibrium that maintains the dormant state and allows the tumor to take off.
Blood markers (VEGF, FGF, placental growth factor, angiostatin, endostatin, and others) and microvessel density are used in the assessment of angiogenesis, but there is no agreement between investigators about the best methods. None of these methods is used in routine practice.
Relationship Between Primary Tumor and Breast Cancer Metastases
Four patterns of metastatic presentation have been proposed. The first pattern, in which metastases grow after removal of the primary tumor, is seen in animal models and believed to be related to suppressive substances released by the primary tumor (angiostatin) that maintain the metastases in dormancy, but they grow rapidly after the primary tumor is removed. The evidence for the existence of this pattern in humans is scant at best. Rapid progression of micrometastases after surgery may be seen in rare cases but may be explained by the postoperative inflammatory state and the dumping of large amounts of cytokines and growth factors in the blood rather than the removal of a putative suppressive substance with the surgical resection of the tumor.
In the second pattern, the metastases grow concomitantly with the primary tumor. The third pattern, known as the occult primary, could be based on metastatic cells that outgrow the primary tumor and suppress its growth by immunologic or other mechanisms such as angiogenesis. The fourth pattern, in which metastases remain dormant for years after removal of the primary tumor, is observed in breast cancer, colon cancer, Ewing sarcoma, and many other tumor types. Again, the mechanism of this prolonged dormancy is unknown. However, recent reports indicate that once metastases become clinically detectable, they display a similar rate of growth that is independent of the number of years of dormancy. This observation is consistent with a model of microscopic dormant metastases that do not expand until sometime within a year of becoming clinically detectable. One explanation is that these metastases were controlled in their new environment by the immune system that was able to overcome their proliferative and angiogenic drive. The regression of metastases after removal of the primary is not known to happen in breast cancer, although it was described in resected renal cell carcinoma.
Disseminated Tumor Cells
Tumors that have acquired the invasive and angiogenic phenotype start shedding malignant cells in the bloodstream (called disseminated tumor cells [DTCs]). DTCs have to overcome multiple resistance mechanisms that lead to the destruction of the majority of them. Only very few can survive this journey and reach distant organs. Once they have colonized these organs, they go through a phase of dormancy that may last for years before they start growing again, causing metastatic cancer that is responsible for 90% of mortality in breast cancer. Breast cancer DTCs have a propensity to relocate to bone. The process is not linear and requires the interaction of DTCs with cells in the destination niche and the immune cells.
The bone marrow offers DTCs two nurturing environments normally occupied by hematopoietic stem cells (HSCs): the osteoblastic (for dormant HSCs) and the vascular (for actively dividing HSCs) niches. Breast cancer DTCs interact through their surface receptor CXCR4 (the same receptor expressed by HSCs) with bone marrow stromal cells such as osteoblasts, endothelial cells, and fibroblasts that express stromal derived factor-1 (SDF-1 or CXCL12), CXCR4 ligand. After relocating to the hematopoietic niche the intruding breast cancer cells may secrete cytokines that suppress hematopoiesis, which may explain the anemia seen in this cancer and others. The predilection to the hematopoietic niche also explains why bone metastases do not arise in the parts of the skeleton that do not harbor red bone marrow. Osteoblasts secrete many cytokines such as angiopoietin-1 and stromal cell factor that help retain HSCs and cancer cells in the niche and by activating the Notch pathway they promote HSC and cancer cell stemness and block their differentiation. New drugs targeting CXCL12 or CXCR4 are used to mobilize HSCs for stem cell transplant but also to “expulse” cancer cells out of the bone marrow. Once out of their protective environment, they become more vulnerable to chemotherapy and/or radiation therapy.
It is possible that complete estrogen deprivation induced by aromatase inhibitors (AIs) has two opposed effects on DTCs in the bone marrow. On one hand, it leads to decreased proliferation or apoptosis of highly estrogen-dependent breast cancer cells. On the other hand, estrogen deprivation induces high bone turnover, resulting in the release of many growth factors embedded in the bone, which may rescue estrogen-deprived cancer cells (insulin-like growth factor) or suppress the immune cells infiltrating and surrounding the tumor (transforming growth factor-β). The net effect is favorable in most patients. This may explain why antiresorptive therapy with bisphosphonates decreases recurrences only in postmenopausal women on AIs. This observation was validated in an animal model.
Tumor Heterogeneity
Molecular Subtypes of Breast Cancer
Oncologists have acknowledged early the heterogeneity of breast cancer based on HR expression. By the beginning of the 1990s, it had become clear that at least three types of breast cancer exist, with distinct clinical behavior and responsiveness to therapy: HR-positive, HER2-positive, and triple-negative breast cancers. The advent of genomic sequencing and the possibility to assess gene expression in cancer ushered in a new era of classifying these diseases based on biologic rather than histologic similarity. On the basis of gene expression profiling, five intrinsic subtypes were identified: the basal-like, the HER2 positive, the luminal A and B, and the normal-like ; subsequent analysis led to the identification of the Claudin low subtype ( Table 24.4 ). This original work led to the development of the PAM50 and PAM50 risk of relapse (PAM50-ROR) assays. The first allows the classification of breast cancers into different intrinsic subtypes, and the second provides important information about their prognosis. In the next decade, other gene expression profile (GEP)-based prognostic assays were developed including the Onco type Dx and MammaPrint. All of these assays generate a score that assigns a low, medium, or high risk of recurrence (PAM50-ROR, Onco type Dx) or good-bad prognosis (MammaPrint) to each individual cancer. Attempts at using a grouping of traditional classifiers such as HR, HER2, and ki67 yielded a good approximation of the intrinsic subtypes (luminal A: HR positive/HER2 negative/ki67 low; luminal B: HR positive/HER2 negative/ki67 high; luminal B: HR positive/HER2-positive; HER2 positive: HR negative/HER2 positive; triple negative: HR negative/HER2 negative) that fell short of fully recapitulating the subtypes defined by GEP.
| Molecular Subtype | Frequency (%) | ER/PR/HER2 | CK5/6 EGFR | Genes of Proliferation | Characteristic Genes | Histologic Grade | TP53 Mutation | Prognosis |
|---|---|---|---|---|---|---|---|---|
| Basal-like | 10–20 | ER− PR− HER2− | + | High | KRT5, CDH3, ID4, FABP7, KRT17, TRIM29, LAMC2 | High | High | Poor |
| HER2-enriched | 10–15 | ER− PR− HER2 + | +/− | High | ERBB2, GRB7 | High | High | Poor |
| Normal breast-like | 5–10 | ER−/+ HER2− | + | Low | PTN, CD36, FABP4, AQP7, ITGA7 | Low | Low | Intermediate |
| Luminal A | 50–60 | ER+ PR+ HER2− | − | Low | ESR1, GATA3, KRT8, KRT18, XBP1, FOXA1, TFF3, CCND1, LIV1 | Low | Low | Excellent |
| Luminal B | 10–20 | ER+/− PR+/− HER2+/− | − | High | ESR1, GATA3, KRT8, KRT18, XBP1, FOXA1, TFF3, SQLE, LAPTM4B | Intermediate/High | Intermediate | Intermediate/poor |
| Claudin-low | 12–14 | ER− PR− HER2− | +/− | High | CD44, SNAI3 | High | High | Poor |
Other tools of genomic analysis have been developed (Onco type Dx, EndoPredict, Breast Cancer Index, and urokinase plasminogen activator and plasminogen activator inhibitor type 1). They provide information about the prognosis and potential benefit from chemotherapy or its ability to lead to pathologic complete response (pCR).
Limitations of Gene Expression Profiling Platforms
GEPs suffer from what we may call static original determinism. The immune system and many stromal and bone marrow–derived cells are involved in carcinogenesis, but they have not been integrated in modern prognostic models (except for the wound-gene signature). GEPs are snapshots of the tumor’s gene expression at one point in time that may not even reflect all the information contained in the tumor due to intratumor heterogeneity. GEPs provide an assessment of interpatient heterogeneity by dividing a group of patients into good and poor prognostic groups. However, it assumes that DTCs in the same patient will carry the same signature as the primary tumor forever and will either progress unavoidably to full-blown metastases (poor prognosis group) or not (good prognosis group). In reality GEPs have had variable success at achieving this task.
The TAILORx trial used the Onco type Dx test to classify node-negative/HR-positive breast cancer patients into three groups (low 0–10, intermediate 11–25, and high >25 recurrence scores [RS]). The first results from the TAILORx trial were recently released regarding patients with low recurrence score (0–10). Of the 10,253 patients enrolled, 1629 (15.9%) had low RS. Five-year distant disease–free survival is 99.3%, and 5-year survival is 98% for these patients. Seventy percent of the patients (70%) had intermediate RS (11–25), and the rest (14.1%) had high RS. The latter group would benefit from chemotherapy, whereas the first group would not. In one way, this is great news for patients with low RS because they will be spared the side effects of chemotherapy. However, the amount of uncertainty remains high for a large subset of patients that fall into the intermediate RS group. The original validation of this test used the cutoff of 18 to define the low recurrence group. Even the “gene dormancy signature” relies on genes of the primary cancer and its predictions rely on the presence or absence of proliferation-promoting genes and they do not do better than the other GEPs.
Intrapatient Heterogeneity and Clonal Evolution
Genetic and epigenetic alterations of breast cancer change over time under the pressure of the microenvironment and the immune system. The treatments used in the adjuvant and neoadjuvant settings add their selection pressure and lead to genetic and epigenetic changes that underlie therapeutic resistance and recurrence. Genetic instability increases over time, leading to the emergence of diverse subclones. This diversity comes at a cost but ultimately offers the cancer a significant survival and dissemination advantage. As tumor cells land in distant organs, cells emerging from some subclones are likely to survive in these new environments and colonize them. Representatives of these subclones can be detected by the analysis of circulating tumor cells in the blood. Colonization may be associated with additional genomic and epigenetic changes. The clonal evolution that began in the primary tumor continues in the secondary metastatic sites leading to further evolution and succession of periods of therapeutic control and failure then ultimately to patient’s death.
Breast cancers vary in their proliferative potential with high proliferation being associated with worse survival outcomes. The drivers for proliferation are different depending on breast cancer subtypes. More than two-thirds of breast cancers express the ER and remain dependent on estrogen for their proliferation for a long time. Estrogen deprivation leads to decreased proliferation and apoptosis of many cells in the tumor mass. This explains the sensitivity of these cancers to estrogen deprivation strategies of long duration. Ultimately, some estrogen-dependent breast cancer cells become resistant to estrogen deprivation by mutating their ER or by activating other proliferation-promoting pathways. The rest of breast cancers do not express ER and depend for their proliferation on activated oncogenes (HER2) or express activated oncogenes in addition to ER. Proliferation of HR-negative breast cancers is cell-autonomous and largely depends on internal drivers. The assessment of proliferation has yet to find reliable and agreed on markers. ki67 has been historically used but the best measurement method and the best cutoff to stratify patients remain to be determined. A cutoff of more than 25% was found to be associated with a greater risk of death compared with lower expression rates in a meta-analysis that included 64,196 patients.
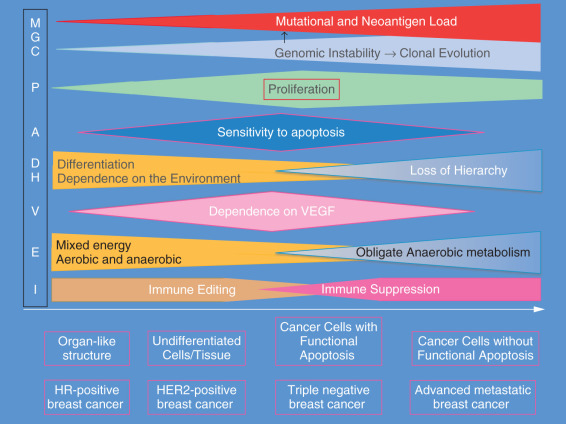
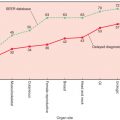
The hallmarks of cancer are expressed to different degrees in different subtypes of breast cancer. At one extreme, estrogen-dependent breast cancers of the luminal A subtype are usually well differentiated (organ-like), and their proliferation rate is low. They are resistant to chemotherapy-induced apoptosis. Their energy metabolism is mixed, aerobic and anaerobic, and they depend little on VEGF. Their mutational and neoantigen loads are low, and their way of immune escape is by immunoediting rather than immune suppression. These cancers may follow an indolent or progressive course. Many of them may remain small, may be detected by screening mammogram, and are responsible of the phenomenon of overdiagnosis seen with the widespread of mammography screening. In TNBC, genomic instability is high and clonal evolution has progressed with multiple clones in the tumor or in distant sites. Proliferation is high, and dependence on external stimuli is minimal if any. Early in the progression of this type, breast cancer cells are highly proliferative and are susceptible to apoptosis (cancer cells with functional apoptotic machinery). Their energy metabolism is becoming more anaerobic and they depend heavily on VEGF. Cells in this subtype are poorly differentiated. Immune escape is achieved by cancer-induced immunosuppression with activation of checkpoints (CTLA4, PD1/PD-L1) and Th2 and M2 polarization of the immune response. These tumors may follow and a systemic model. However, it is clear that there are many possible combinations of different hallmarks of cancer that may lead to the different courses described in the previous section ( Fig. 24.3 ).
Therapeutic Strategies for the Management of Invasive Breast Cancer
Strategies that have been developed to manage breast cancer span the spectrum of the disease from its inception to its diagnosis and treatment than to the posttreatment phase and take advantage of the weaknesses of the cancer. Initiation and progression of breast cancer require (1) a strong estrogenic drive and/or (2) genetic and epigenetic alterations; (3) loss of intracellular control and decreased apoptotic predisposition; (4) weak immune surveillance and chronic inflammatory milieu; and (5) a permissive microenvironment. Inherited factors act by magnifying one or more of the previous disturbances.
Surgery leads to the physical elimination of the tumor and radiation and chemotherapy induce DNA damage or interrupt the cell cycle, which would overwhelm cancer cell repair mechanisms leading to active cell death. In all of these cases, it is possible that the end result is the eradication of cancer cells. Hormone blockade or estrogen depletion may lead to the induction of dormancy of tumor initiating cells. This strategy does not lead to complete elimination of cancer cells that may wake up if the treatment is interrupted. Angiogenesis inhibitors did not prove effective in the adjuvant setting. The failure of targeting angiogenesis in the adjuvant/neoadjuvant setting is one of the most important lessons in the late 2000s that suggest clearly that angiogenesis is not an important mechanism for the maintenance of DTCs or micrometastatic disease. Targeted therapy resulted in a major shift in the treatment of HER2-positive cancer. Recent discoveries suggest that this modality works through activation of the immune system rather than the interruption of the oncogenic drive of HER2. The recently rediscovered immune therapy may induce either one of these outcomes—eradication or dormancy—and is a promising method for opening new horizons in the treatment of cancers, including breast cancer.
The most important principle in clinical management of breast cancer patients is the need for close collaboration among the members of a multidisciplinary team that includes surgeons, medical oncologists, radiation oncologists, radiologists, pathologists, and representatives of different support services. Patients are stratified according to the TNM staging. The TNM stage is one of the first prognostic factors that would determine the prognosis and the type of treatment. Early-stage breast cancers are those with stage I, IIA, and IIB (T2N1). Locally advanced breast cancers are those with stage IIB (T3N0) and stage IIIA though IIIC. This chapter focuses mainly on the multidisciplinary management of early invasive breast cancer. For detailed discussion of the management of locally advanced breast cancer see Chapter 65 .
Surgical Treatment
The main stay of surgical treatment of breast cancer is the complete resection of the cancer en bloc with a rim of normal tissue while preserving function and cosmesis. Progress in understanding the biology of breast cancer and improvement of surgical techniques led a revolutionary reversal of the increasingly mutilating surgeries that were performed in the first half of the 20th century. Radical mastectomy was replaced by modified radical mastectomy then lumpectomy (breast conservation surgery [BCS]) with radiation therapy (RT) (BCS + RT + breast conserving therapy [BCT]). This change became possible with the increased usage of screening mammography and the detection of tumors at very small sizes.
National Surgical Adjuvant Breast and Bowel Project (NSABP) B-4 was a seminal trial that propelled BCS forward in the 1970s. It was the first trial to randomize patients with clinically negative or positive lymph nodes to radical mastectomy (RM) or total mastectomy (TM) with RT. The trial accrued its 1665 patients between 1971 and 1974. They were randomized to the planned procedures and were followed for up to 6 years (average of 36 months). At the time of the publication of the study results, 14% of those with clinically negative lymph nodes who underwent TM (without axillary lymph node dissection [ALND]) had a regional axillary lymph node failure. Patients with clinically negative lymph nodes who underwent RM or TM + RT had no axillary recurrences. Patients who had locoregional failure were rescued with ALND. Disease-free survival did not significantly differ between patients with clinically node-negative axilla on the three arms including those who had a locoregional recurrence and were subsequently rescued with surgery.
Veronesi and coworkers demonstrated equivalence between BCT and RM by showing that all-cause mortality was practically the same (BCT 41.7%, RM 41.2%) in the two groups after 20 years of follow-up. The slight increase in locoregional recurrence in the BCT group compared with RM (8.8% vs. 2.3%) did not affect long-term survival. The need for RT was explored by NSABP B-6, which demonstrated that BCS alone is associated with ipsilateral breast tumor recurrence (IBTR) in 39.2% compared with 14.3% when RT was added to BCS. These seminal studies established the role of BCT that became the predominant form of surgery performed in the developed countries. The Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) reviewed the data on 10,801 women in 17 randomized trials and concluded that RT reduces the risk of locoregional recurrence after BCS by half. Several observational studies have shown that BCT offers equivalent—and even superior—long-term survival to patients compared with TM. However, it was argued that the observational studies by their nature carry implicit and explicit biases that may be responsible for these results.
Patients who are not eligible for BCT include the following: (1) pregnant women, (2) patients who have had prior RT, (3) patients with multicentric disease or diffuse calcifications on mammography, (4) those with large tumors relative to the breast size, and (5) persistent positive margins after reexcision. Patient preference should be added.
Modified radical mastectomy with axillary lymph node sampling of level I and/or II may be needed to remove all cancer. Otherwise, simple mastectomy is performed if the lymph nodes are clinically negative.
Lymph nodes evaluation is performed with fine-needle aspiration or core needle biopsy if the nodes are clinically positive or with sentinel lymph node biopsy (SLNB) if they are clinically negative. If the SLNB is negative for cancer involvement, no further action is undertaken; if it is positive, ALND is performed. On the basis of the American College of Surgeons Oncology Group trial Z-11, forgoing ALND in patients with clinical T1–2N0 and one or two positive sentinel LNs after undergoing BCS is oncologically safe and is associated with less morbidity. A 10-year follow-up of this study showed no significant difference between the group that received the ALND and the observation group with locoregional recurrence-free survival at 93.2% for the ALND arm and 94.1% for the SNB arm.
Debate regarding margin width continued for years and led to many surgical reexcisions—many of which were not necessary—until a working group of experts from the Society of Surgical Oncology and the American Society for Radiation Oncology met and reviewed the evidence in 2014. They reviewed data from 33 eligible studies published between 1965 and 2013 and included 28,162 patients, of whom 1506 had an IBTR. The conclusion of this meta-analysis was that a “positive margin, defined as ink on invasive cancer or ductal carcinoma in situ (DCIS), is associated with at least a 2-fold increase in IBTR.” RT, systemic therapy, or favorable biology do not nullify this increased risk.
Radiation Therapy
RT is based on the induction of massive DNA damage in cancer cell genomes that lead to apoptosis. RT is a locoregional treatment modality that helps limit the extent of surgery, although RT works better in the context of minimal residual disease. The EBCTCG and the St. Gallen International Expert Consensus Group recently updated their findings/guidelines regarding RT.
- 1.
Partial mastectomy and whole breast irradiation (WBI) are equivalent to total mastectomy.
- 2.
Patients who undergo BCS are offered WBI because it decreases the risk of LRR and IBTR and may lead to improved survival. The EBCTCG overview showed that for every four local recurrences prevented, one breast cancer death is avoided at 15 years of follow-up after diagnosis.
- 3.
Patients who undergo mastectomy with negative SLNB do not require RT. However, some patients who underwent mastectomy still benefit from RT if they had a tumor greater than 5 cm, chest wall invasion, positive macrometastatic sentinel node biopsy but no axillary dissection, patients with one to three involved nodes and adverse pathology, involvement of four or more lymph nodes, or in presence of extranodal extension.
- 4.
Regional nodal irradiation (RNI) is not indicated if lymph nodes are negative. If the lymph nodes are positive, the addition of RNI to whole breast RT decreases IBTR without changing long-term survival in an RCT. An observational study showed that radiating the internal mammary LNs (IMLNs) might be associated with survival advantage if IMLNs were positive.
- 5.
Women older than 70 with HR-positive stage I breast cancer may forgo RT and be offered tamoxifen after lumpectomy with excellent long-term results. Although local recurrence was higher in the no RT group there was no negative effect on survival.
- 6.
Hypofractionated RT using 15 to 16 fractions is considered standard of care by many experts.
- 7.
Partial breast irradiation (PBI) is at least equivalent to WBI in selected patients and it may be associated with lower nonbreast cancer and overall mortality (up to 25% relative decrease in mortality).
Medical Treatment
The major lesson learned from the surgical management of breast cancer since the 1960s was that the type of local management does not affect long-term outcome of the disease that depends on the presence of DTCs/micrometastases outside of the breast. This realization paved the way to systemic therapy with the goal to eradicate DTCs and micrometastases. Major advances in systemic therapy have benefited from better understanding of the biology of breast cancer.
Several strategies have been used to eradicate the hidden reservoir of DTCs and thus prevent recurrence of the disease.
- 1.
The first strategy was based on endocrine manipulations to induce estrogen deprivation. It is possible that this strategy leads to maintaining tumor dormancy instead of its complete eradication.
- 2.
The second strategy was the use of chemotherapy to induce cytotoxicity and cell death. Many chemotherapeutic drugs have been tried since the 1960s to achieve this goal. As we will see, some of the effects of chemotherapy may be related to its immunomodulatory effect.
- 3.
The third strategy emerged in the 1990’s after the discovery of oncogenes and tumor suppressor genes. This was the golden age of targeted therapy that allowed major advancements to cure certain types of breast cancer through the targeting of activated oncogenes.
- 4.
Targeting tumor angiogenesis did not result in any significant improvement of outcomes in the adjuvant/neoadjuvant setting.
- 5.
Finally, we are transitioning now to the era of immunotherapy.
Chemotherapy
The success of chemotherapy in childhood leukemia ushered in a new era for the treatment of cancer. Many of the drugs that had shown efficacy in hematologic malignancies were tried in solid tumors including breast cancer. The success of cyclophosphamide, methotrexate, and 5-fluorouracil (CMF) designed by Bonadonna was followed by other combination regimens with equal or superior efficacy ( Tables 24.5 and 24.6 ).
| Generation | Benefit | Regimens With Substantial Evidence Base |
|---|---|---|
| First | 35% reduction in breast cancer mortality compared with no adjuvant chemotherapy | CMF ×6, AC ×4, FEC50 ×6 |
| Second | 20% reduction in breast cancer mortality compared with first generation regimen | FEC100 ×6, CAF ×6, FAC ×6 |
| AC ×4, T ×4 (every 3 wk) | ||
| DC ×4, E ×4-CMF ×4 | ||
| Third | 20% reduction in breast cancer mortality compared with second generation regimen | FEC ×4, D ×3, FEC ×4 weekly T ×8 |
| Concurrent DAC | ||
| Dose-dense AC ×4, T ×4 | ||
| AC ×4 weekly paclitaxel | ||
| AC ×4 docetaxel (every 3 wk) |
| Generation | Comparison (Reference) | Nodal Status | Patient N | Median Follow-Up (Years) | HR for DFS | HR for OS |
|---|---|---|---|---|---|---|
| First | CMF vs. no chemo | + | 386 | 28.5 | 0.71 ( p = .005) | 0.79 ( p = .04) |
| CMF + Tam vs. Tam (B20) | − | 2306 | 5 | 0.65 ( p = .001) | 0.64 ( p = .03) | |
| AC vs. CMF (B15) | + | 2194 | 3 | p = .5 a | p = .8 a | |
| AC vs. CMF (B23) | − | 2008 | 5 | p = .9 a | p = .4 a | |
| FEC50 + Tam vs. Tam | + | 457 | 9.4 | 0.46 ( p = .0008) | 0.65 ( p = .07) | |
| Second | FEC100 vs. FEC50 | + | 546 | 5.6 | 0.63 ( p = .02) | 0.45 ( p = .005) |
| ACx4-T ×4 vs. AC ×4 (C9344) | + | 3121 | 5.8 | 0.83 ( p = .002) | 0.82 ( p = .006) | |
| ACx4-T ×4 vs. AC ×4 (B28) | + 0–3 | 3060 | 5.4 | 0.83 ( p = .006) | 0.93 ( p = .46) | |
| DC ×4 vs. AC ×4 | + | 1016 | 7 | 0.74 ( p = .033) | 0.69 ( p = .032) | |
| Ex4-CMF ×4 vs. CMF ×6/CMF ×8 | + − | 2391 | 4 | 0.69 ( p < .001) | 0.67 ( p < .001) | |
| Third | DAC vs. FAC | + | 1491 | 10.3 | 0.80 ( p = .004) | 0.74 ( p = .002) |
| DAC vs. FAC | − | 1060 | 6.4 | 0.68 ( p = .01) | 0.76 ( p = .29) | |
| FEC-D vs. FEC | + | 1099 | 7.8 | 0.85 ( p = .036) | 0.75 ( p = .007) | |
| FEC-weekly T vs. FEC | + | 1246 | 5.5 | 0.77 ( p = .022) | 0.78 ( p = .11) | |
| FAC-weekly T vs. FAC | − | 1925 | 5.3 | 0.73 ( p = .04) | 0.79 ( p = .31) | |
| Q3 vs. q2wk ACT | + | 2005 | 5.8 | 0.80 ( p = .01) | 0.85 ( p = .04) | |
| AC-T vs. AC-weekly T | + | 4954 | 12.1 | 0.84 ( p = .011) | 0.87 ( p = .09) | |
| AC-T vs. AC-D | 0.79 ( p = .001 | 0.86 ( p = .054) | ||||
| AC-D vs. DAC | + | 5351 | 6.1 | 0.80 ( p = .001) | 0.86 ( p = .09) | |
| AC-D vs. AD | 0.83 ( p = .01) | 0.83 ( p = .03) |
Related posts:
 Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
 Mesenchymal Neoplasms and Primary Lymphomas of the Breast
Mesenchymal Neoplasms and Primary Lymphomas of the Breast
 Breast Conservation Therapy for Invasive Breast Cancer
Breast Conservation Therapy for Invasive Breast Cancer
 Radiation Complications and Their Management
Radiation Complications and Their Management
 Immunologic Approaches to Breast Cancer Therapy
Immunologic Approaches to Breast Cancer Therapy
 Delayed Diagnosis of Symptomatic Breast Cancer
Delayed Diagnosis of Symptomatic Breast Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


