Fig. 5.1
T cell activation requires recognition of the antigen and costimulatory signals. Inflammation generated by tissue damage or infections activates antigen-presenting cells (APCs) and stimulates the expression of costimulatory molecules, such as CD80/CD86. Presentation of the antigen to the T cell receptor (TCR), in the context of major histocompatibility complex (MHC) molecules and CD80/CD86 that interact with CD28, stimulates the expansion and differentiation of naïve T cells (top panel). Resting APCs express few or no costimulatory molecules and fail to activate T cells, this leads to anergy (middle panel). CTLA-4 is a coregulatory molecule that binds CD80 and CD86 and is upregulated on activated T cells. CD80/CD86-CTLA-4 interactions inhibit T cell responses and mediate tolerance
Although costimulatory molecules were initially identified as stimulators of T cell responses, some costimulatory (coregulatory) receptors inhibit T cells [1]. Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) is a CD28 homolog that also binds CD80 and CD86. Nevertheless, CTLA-4 expression is inducible after T cell activation, and is involved in the induction and maintenance of tolerance, as its ligation inhibits IL-2 production and blocks cell cycle progression [1].
After the discovery of homologs of CD28/CTLA-4 and their ligands, many other coregulatory molecules have been identified, some of which include the inducible T cell costimulator (ICOS or CD278) with its ligand CD275 (ICOS-L, B7h, B7-RP), the inhibitory programmed death-1 (PD-1, CD279) with its ligands PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273), and the B- and T-lymphocyte attenuator (BTLA, CD272) which binds the herpesvirus entry mediator (HVEM). BTLA is an additional receptor of the immunoglobulin superfamily that negatively regulates T cell activation. In addition, HVEM interacts with another negative regulator of T cells, CD160. Recent studies of the lymphocyte activation gene-3 (LAG-3, CD223) suggest that this molecule also plays an important role in the regulation of T cell responses. Moreover, the T cell immunoglobulin domain and mucin domain-3 (TIM-3), with its ligand galectin-9, are involved in terminating Th1 cell responses and establishing tolerance [2, 3].
T cells that recognize antigen in the absence of costimulation either fail to respond and undergo cell death or enter a state of unresponsiveness, a phenomenon known as anergy. Thus, costimulation is a key factor in the outcome of T cell interactions with the antigen. Significant efforts have been undertaken to characterize costimulatory molecules in order to augment antitumor responses; recent evidence has demonstrated the importance of coregulatory molecules in the inhibition of immune responses. Thus, interfering with these regulatory pathways has gained interest as a potential strategy for cancer therapy [4].
5.3 T Cell Anergy
T cell anergy induces peripheral tolerance; this mechanism protects the host from autoimmune diseases. In addition, anergy has been suggested to play an important role in the induction of T cell dysfunction in cancer patients. T cell anergy is a tolerance mechanism in which, after antigen encounter, the T cell is intrinsically and functionally inactivated [5]. The cell remains alive in this hyporesponsive state for an extended period of time. Anergic T cells neither produce nor respond to proliferative signals and are unable to exert effector functions, such as cytolysis or cytokine secretion. A characteristic of anergy is that it must be cell autonomous, which distinguishes this process from immunoregulation mediated through other regulatory cells, such as regulatory T cells (Tregs) [5, 6].
There are at least five distinct sets of circumstances that lead to T cell anergy [5, 7]: (1) TCR ligation in the absence of full costimulation; (2) exposure to partial agonists, peptides with minor sequence differences from native agonist antigenic peptides that exhibit reduced avidity for TCR ligation; (3) full signaling without IL-2 receptor-driven cell division; (4) TCR ligation in the presence of IL-10 or transforming growth factor-β (TGF-β); and (5) anergy induced through CTLA-4 or other coregulatory molecules (Fig. 5.1).
Thus, anergy is the consequence of factors that negatively regulate proximal TCR-coupled signal transduction, together with active transcriptional silencing, which is reinforced through epigenetic modifications [8]. This state of nonresponsiveness is molecularly distinct from T cell exhaustion. T cell anergy is induced upon the first encounter with the antigen and is quickly initiated, in contrast with T cell exhaustion, which is progressive. Gene expression profiles show that anergy and exhaustion are partially distinct. Genes, such as Rnf128 (Grail), Egr2, and Egr3, are upregulated in anergic but not in exhausted T cells, whereas NFAT is upregulated under both conditions [9]. The detailed characterization of the differences between anergy and T cell exhaustion will have important implications for therapeutic interventions in immune-mediated diseases and chronic infections.
5.3.1 T Cell Anergy in Cancer
Anergy has been proposed to play a role in the impairment of T cell function in human cancers. Tumor cells are poor APCs, as these cells express antigens on MHC class I molecules but do not express costimulatory molecules to provide a second signal for full T cell activation; thus, tumor-infiltrating lymphocytes (TILs) are rendered anergic [10]. In addition, immature myeloid-derived dendritic cells (mDCs), plasmocytoid dendritic cells (pDCs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) potentially induce anergy in TILs [8, 11, 12]. Several studies have shown that human tumor cells, mDCs, pDCs, MDSCs, and TAMs express high levels of coregulatory molecules, such as PD-L1, PD-L2, ICOS-L (B7-H2, CD275), and B7-H3 (CD276), indicating a poor costimulatory and a high inhibitory anergy-promoting environment. Evidence that cancer induces T cell anergy comes from studies where the transfection of CD80 in tumor cells or the blockage of the B7 family coregulatory molecules results in reduced tumor growth or tumor rejection in mice models [2, 11–14].
Analysis of the functional state of TILs has demonstrated that these cells are characterized by impairment of cytolytic activity, decreased cytokine secretion, reduced expression of IL-2Rα (CD25), and diminished activation of extracellular signal-regulated kinase (ERK) after TCR activation. Thus, T cell anergy occurs in the tumor microenvironment of some human tumors [14–16]. Nevertheless, direct evidence that T cell anergy occurs in cancer has been difficult to obtain due to the lack of surface markers to identify anergic T cells [8].
Based on mouse tumor models, the induction of antigen-specific T cell anergy has been suggested to be an early event in the progression of tumors, which occurs in the equilibrium phase of immunoediting, before immunosuppression takes place in advanced tumors (escape phase) [10, 17]. However, Klein et al. showed that highly immunogenic tumors evade immunosurveillance due to antigen overload and an insufficient number of tumor-specific T cells, resulting in the exhaustion of the immune cells [18]. Thus, from a temporal perspective, T cell anergy predominantly occurs during the early stages of tumor progression, whereas T cell exhaustion might play a crucial role in T cell dysfunction during the late stages of cancer [10].
5.4 T Cell Exhaustion
T cell exhaustion has been defined as a stage of T cell differentiation where T cells have poor effector functions, sustained coregulatory receptor expression, and a transcriptional state distinct from that of functional effector or memory T cells [19]. Originally, this phenomenon was identified in chronic viral infections in mice and later in chronic viral infections in humans, e.g., human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV) [19–22]. Chronic bacterial and parasitic infections have been demonstrated to induce T cell exhaustion; also, cancer has been suggested to induce a similar phenomenon [20, 23].
During chronic infections, antigen-specific CD8+ T cells initially acquire effector functions, but gradually become less functional as the infection progresses. The dysfunction of exhausted T cells is hierarchical, showing the initial loss of properties, such as cytotoxic activity, proliferative potential, and interleukin 2 (IL-2) synthesis; followed by diminished tumor necrosis factor-alpha (TNF-α) secretion and subsequent loss of interferon-gamma (IFN-γ) production during the late stages of exhaustion. Finally, during the most extreme stages of exhaustion, deletion of T cells occurs through apoptosis [19, 24] (Fig. 5.2). Like CD8+ T cells, CD4+ T cells also lose function during chronic infections; however, there is little information about the mechanisms of exhaustion in this T cell subpopulation [19].
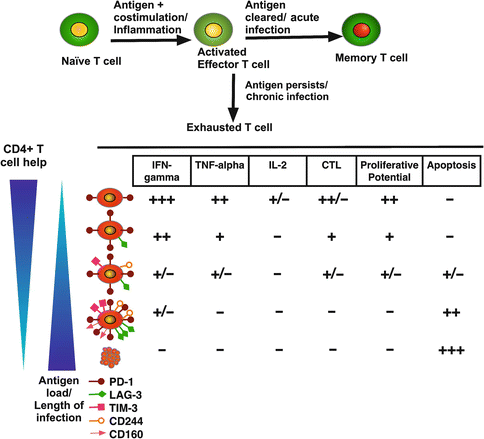
Fig. 5.2
T cell exhaustion during chronic inflammation. In an acute inflammatory process, naïve T cells are primed by an antigen, costimulatory molecules, and cytokines that promote differentiation into effector T cells. After clearance of the antigen and once inflammation is resolved, a subset of effector T cells differentiates to become memory cells. During chronic processes, such as viral infections, the antigen persists, and T cells go through several stages of dysfunction, losing effector functions (cytolysis and secretion of cytokines) and proliferative potential in a hierarchical manner. Finally, deletion of T cells by apoptosis occurs. As antigen load increases or CD4+ T helper subpopulation decreases, T cells become more exhausted. Expression of coregulatory receptors is correlated with the level of exhaustion. The scale of each activity is presented from high (+++) to low (−)
Exhausted T cells possess a molecular profile that is distinct from those of memory, effector, and anergic T cells [9]. First, many membrane inhibitory receptors are upregulated, for instance, PD-1, LAG-3, and TIM-3. Second, transcription of genes encoding molecules involved in TCR signaling (such as Lck and NFATc) and cytokine receptors (IL7 and IL-15 receptors) are downregulated. Third, the pattern of genes involved in chemotaxis, migration, and adhesion is changed. Fourth, there is an altered pattern of differentiation compared with memory or effector T cells. Finally, exhausted T cells present deficiencies in translational, metabolic, and bioenergetic processes, such as the Krebs cycle [9].
5.4.1 Mechanisms for Inducing T Cell Exhaustion
Immunoregulation is critical in T cell exhaustion. Coregulatory receptors play a key role in many aspects of adaptive immunity, including self-tolerance, prevention of autoimmunity, and cancer. The mechanisms of regulation through coregulatory receptors have not been characterized in detail; nevertheless, several studies suggest that these receptors attenuate T cell responses in many ways. Accumulating evidence highlights the pivotal role of the PD-1/PD-L1 pathway in maintaining an immunosuppressive tumor microenvironment. This pathway has been proposed to be the most important coregulatory pathway involved in T cell exhaustion [25, 26].
A transmembrane receptor of the Ig superfamily, PD-1 (CD279), is upregulated in mice chronically infected with lymphocytic choriomeningitis virus (LCMV) [25]. PD-1 interacts with its ligands PD-L1 (B7-H1, CD274) or PD-L2 (B7-DC, CD273), which are members of the B7 family [26]. PD-1 is rapidly upregulated on activated T cells; then, after antigen clearance, the expression of this receptor is reduced on effector T cells. Upon subsequent antigen stimulation, effector T cells show upregulated PD-1 expression. Thus, the continuous stimulation of T cells (naïve or effector) during chronic infections induces the accumulation of PD-1+ T cells [19]. High levels of PD-L1 expression on APCs (or tumor cells) might sustain PD-1 expression on T cells, impair T cell effector maturation, and allow the progression of chronic infection [27–29].
Studies in mouse tumor models demonstrated that the inhibition of PD-L1 or PD-1 using blocking monoclonal antibodies (mAbs) increases the cytolytic activity of CD8+ T cells and reverses T cell dysfunction [30, 31]. Subsequently, Barber et al. showed that the inhibition of PD-1 using anti-PD-1 mAbs in chronically infected mice enhances the proliferation and effector functions of exhausted T cells [25]. Since the publication of these seminal reports, many other studies have shown that PD-1 with its ligand (PD-L1) is crucially involved in T cell exhaustion in chronic human pathogen infections and cancer [21–24, 32–34].
In addition to PD-1, many other cell surface inhibitory receptors also participate in T cell exhaustion. These coregulatory receptors regulate distinct cellular functions. For instance, PD-1 pathway affects T cell survival and proliferation, whereas LAG-3 affects cell cycle progression, but has less influence on apoptosis [19]. Several receptors belonging to the tumor necrosis receptor family are upregulated in exhausted T cells, such as Fas, TNF-R, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors; hence, these death receptors have been implicated in the induction of exhaustion, as T cells might become prone to activation-induced cell death (AICD) [19, 35, 36].
TIM-3 is an inhibitory molecule that downregulates effector Th1 responses; upregulation of this molecule has been found in HIV-specific and HCV-specific CD8+ T cells in patients with progressive HIV and HCV infections, respectively. Importantly, the coexpression of TIM-3 and PD-1 has been associated with severe CD8+ T cell exhaustion in terms of the proliferation as well as secretion of effector cytokines, such as IFN-γ, TNF-α, and IL-2 [19]. Interestingly, CD8+ T cells expressing both coregulatory receptors also produce the suppressive cytokine IL-10 [37].
Remarkably, functional effector T cells express coregulatory receptors during activation; however, as indicated above, the prolonged and increased expression of multiple coregulatory receptors is a key feature of CD4+ and CD8+ T cell exhaustion. However, exhausted T cells do not necessarily coexpress all of the coregulatory molecules. The pattern as well as the level of expression of coregulatory receptors simultaneously expressed in the same CD8+ T cell might considerably influence the severity of dysfunction [38].
Several factors, such as duration of the infection, level of antigen exposure, availability of CD4+ T cell help, and the type of APCs that present the antigen, have been implicated in the severity of T cell exhaustion. Ligand availability for coregulatory receptors could also influence the degree of exhaustion, as well as environmental factors such as the presence of immunoregulatory cytokines [19]. In chronic viral infections, IL-10 expression is associated with T cell dysfunction [38, 39]. In addition, TGF-β has also been linked to exhaustion in chronic infections in humans [40, 41]; nevertheless, the mechanisms underlying IL-10 and TGF-β-mediated T cell exhaustion are unclear. Interestingly, both cytokines are secreted by several human tumors [42, 43].
5.4.2 Identification of Exhausted T Cells
Exhausted T cells show a poorly differentiated phenotype (CD27hiCD28loCD57loCD127loCCR7-CD45RA+ or CD27+CD45RO+) correlated with T cell dysfunction. Although PD-1 upregulation in T cells was initially considered as a hallmark of T cell exhaustion, this molecule is upregulated along with activation markers, such as CD38 or HLA-DR [44]. In healthy adults, the percentage of PD-1+ cells varies from 40 to 80 % of (CCR7+/−CD45RA−) memory T cells; remarkably, these cells do not exhibit characteristics of exhaustion [45]. Thus, PD-1 is also associated with T cell activation and differentiation.
Many cell surface coregulatory receptors are expressed in exhausted T cells. LAG-3, TIM-3, CD244 (2B4), CD160, CTLA-4, and the recently described B- and T-lymphocyte attenuator (BTLA) are coexpressed in antigen-specific CD8+ T cells during chronic infection. The pattern and level of coregulatory receptors simultaneously expressed in the same CD8+ T cell considerably influence the severity of dysfunction [38]. However, depending on the chronic infections or cancer, exhausted T cells might express a different pattern of coregulatory molecules.
Genomic strategies have provided a molecular profile for exhausted T cells. These genomic studies support the notion that T cell exhaustion represents a particular state of differentiation, different from that of effector or memory T cells [9, 19].
Several transcriptional pathways have been associated with T cell exhaustion. The increased expression of transcriptional repressor Blimp-1 is associated with the upregulation of many coregulatory receptors (PD-1, LAG-3, CD160, and CD244). In addition, the transcription factor NFATc1 (NFAT2) is also upregulated but shows a dysregulated function [9]. The transcription factor T-bet also plays a role in protection against T cell exhaustion, as T-bet promotes terminal differentiation after acute infection, and the increased expression of this transcription factor inhibits the expression of coregulatory receptors during chronic viral infection. T-bet expression is downregulated through persistent antigenic stimulation, resulting in T cell exhaustion [46].
5.5 T Cell Exhaustion in Cancer
Cancer and chronic viral infections have been thought to share similar mechanisms in establishing high antigen load and an immunosuppressive environment. However, there is a fundamental difference between both diseases: viral antigens are exogenous and extremely immunogenic, whereas tumor antigens are self-molecules that are weakly immunogenic. Thus, compared with tumor-specific T cells, virus-specific T cells are more frequent and easily detectable, facilitating the ease in identification, phenotypic characterization, and isolation of T cells [10]. However, in the tumor microenvironment, infiltrating T cells become dysfunctional and show reduced effector functions. Several reports suggest that PD-L1 expression on tumor cells plays an important role in tumor-induced T cell dysfunction. PD-L1 membrane expression has been observed using immunohistochemistry on many human tumors, such as melanoma, lung, larynx, colon, breast, cervix, and stomach [26]. In breast, esophageal, gastric, and renal carcinomas, the increased expression of PD-L1 on the surface of tumor cells is strongly associated with poor prognosis [26, 47]. Thus, T cell exhaustion has been proposed as a mechanism for inducing T cell dysfunction through the PDL-1/PD-1 pathway. However, as previously indicated, PD-1 expression cannot be considered as the sole marker of T cell exhaustion in chronic diseases and cancer; hence, other markers must be considered.
In human metastatic-melanoma lesions, TILs show upregulation of PD-1 expression, accompanied with reduced production of IFN-γ TNF-α, and IL-2. Both tumor-infiltrating CD8+ T cells, particularly MART-1-specific, and tumor-infiltrating CD4+ T cells show significantly higher levels of PD-1 expression than CD8+ and CD4+ T cells from peripheral blood and normal tissues from cancer patients. In addition, a large proportion of CD8+ T cells from TILs were PD-1+CTLA-4+ cells compared with normal tissues and blood. Furthermore, PD-1+CD8+ cells from TILs lacked CD25 as well as IL-7RA expression, suggesting that these cells were unable to proliferate, produce effector cytokines, and differentiate into memory cells [48]. PD-1+NY-ESO-1-specific CD8+ T cells, from patients with advanced melanoma, upregulate TIM3 expression and are more dysfunctional than TIM3–PD-1+ and TIM3–PD-1−NY-ESO-1-specific CD8+ T cells, producing less IFN-γ, TNF-α, and IL-2 [49].
Derré et al. showed that tumor antigen (Melan-A/Mart-1)-specific CD8+ T cells express high levels of BTLA and are susceptible to functional inhibition through its ligand HVEM [50]. In addition, Baitsch et al. recently showed that in melanoma, tumor antigen-specific CD8+ T cells with effector phenotypes simultaneously express four or more of the inhibitory receptors BTLA, TIM-3, LAG-3, KRLG-1, 2B4, CD160, PD-1, or CTLA-4 [51]. Moreover, tumor antigen-specific CD8+ T cells present a large variety of genes with a similar genetic profile as that of exhausted T cells from chronic viral infections [52]. Taken together, these reports show that tumor antigen-specific CD8+ T cells are exhausted in melanoma patients.
Additional evidence for T cell exhaustion in other cancers comes from studies in patients with ovarian cancer. Matsusaki et al. reported that NY-ESO-1-specific CD8+ T cells, from the peripheral blood of patients with ovarian cancer, show impaired effector functions along with coexpression of the inhibitory molecules LAG-3 and PD-1. The expression of LAG-3 and PD-1 on the surface of CD8+ T cells is upregulated through IL-10, IL-6, and tumor-derived APCs. In addition, LAG-3+PD-1+CD8+ T cells are deficient in IFN-γ/TNF-α secretion compared with LAG-3+PD-1− or LAG-3−PD-1− subsets [53].
In hepatocellular carcinoma, PD-1+CD8+ T cells are higher in tumor tissues than in non-tumor tissues, presenting decreased proliferative abilities as well as effector functions, as demonstrated by reduced granule and cytokine expression compared with PD-1−CD8+ T cells [54]. Nevertheless, no other marker of T cell exhaustion was analyzed.
PD-L1 expression is upregulated in Hodgkin lymphoma (HL) and several T cell lymphomas, but not in B cell lymphomas. In addition, PD-1 is upregulated in TILs as well as peripheral blood T cells from HL patients and the blockade of the PD-1 pathway restores IFN-γ production in T cells [55]. Moreover, LAG-3 is expressed on TILs from patients with this malignancy [56]. Taken together, these reports suggest that TILs from patients with HL are exhausted.
In patients with chronic lymphocytic leukemia (CLL), CD8+ and CD4+ effector T cells show the increased expression of CD244, CD160, and PD-1, with the expansion of the PD-1+ BlimpH1 subset. CD8+ T cells from CCL patients show defects in proliferation and cytotoxicity, but with increased production of IFN-γ and TNF-α, normal production of IL-2, and increased expression of T-bet. Thus, although CD8+ T cells show features of T cell exhaustion, these cells retain the ability to produce cytokines [57]. However, head and neck cancers that are positive for human papillomavirus (HPV) present a high infiltration of PD-1+ T cells, and the numbers of PD-1+ cells are positively associated with a favorable clinical outcome. Nevertheless, these PD-1+ T cells express activation markers, 50 % of this population lack TIM-3 expression, and are functional after the blockade of the PD-1/PD-L1 pathway, suggesting that PD-1+ T cells are activated rather than exhausted [58].
Interestingly, Haymaker et al. proposed that PD-1highCD8+ T cells in cancer patients are not exhausted [59]. This hypothesis is based on the observation that CD8+ T cells from the TILs of melanoma patients recover their proliferative potential ex vivo, despite expressing high levels of PD-1. These TILs mediate antitumor responses upon adoptive transfer into patients [60, 61]. Under this hypothesis, infiltrating and peripheral blood CD8+ T cells, expressing PD-1, BTLA, along with other coregulatory receptors, are not exhausted. Instead, these cells are highly activated effector-memory cells T cells that can be stimulated through immunotherapy [59]. Nevertheless, these observations have been primarily achieved in melanomas. In other cancers, the reduced proliferative and effector capacities persist, even after stimulation, and immunotherapeutic strategies have failed to induce potent antitumoral responses [53, 57, 62, 63].
Some of the phenotypic, functional, and molecular changes that occur in T cells during chronic infections are exhibited in TILs as well as peripheral blood T cells from several cancer types. The initial aim of tumor immunotherapy was to prevent anergy and tolerance towards tumor antigens. However, the efficacy of this strategy is potentially limited by T cell exhaustion [10]. Interestingly, Hailemichael et al. showed that in mice vaccinated with gp100 melanoma peptide, the persisting tumor antigen at vaccination sites induces the sequestration of CD8+ T cells, resulting in the dysfunction and death of these cells [63].
PD-1 blockage results in the recovery of T cell effector functions in vitro and in animal models in several tumors, thus significantly enhancing antitumor immunity [30, 31, 49, 64]. This knowledge has been translated into several clinical trials [34, 65]. Recently, Brahmer et al. showed that the antibody-mediated blockade of PD-L1 induced durable tumor regression along with prolonged disease stabilization in patients with selected advanced cancers, including non-small cell lung cancer [65]. Thus, understanding T cell exhaustion in cancer will contribute to the advancement of tumor immunotherapy.
5.5.1 A Particular Case: T Cell Exhaustion in Lung Cancer Patients
Lung cancer is the leading cause of cancer-related mortality in developed countries and the second leading cause of death in countries with emerging economies. Lung cancer is one of the most commonly diagnosed cancers worldwide, representing 13 % of all cancer cases and approximately 18 % of all cancer deaths [66]. Some reports show that the presence of TILs with memory phenotype in lung cancer is predictive of a favorable clinical outcome [67–69]. Also, it has been shown that CD8+ T cell subpopulation is decreased in the pleural compartment with respect to peripheral blood from lung cancer patients, whereas the CD4+ T cell subpopulation is increased [70, 71].
Both in TIL and in the pleural compartment, CD8+ T cells are functionally impaired and are poorly responsive or unresponsive to several T cell-activating stimuli, even though memory cells infiltrate lung tumors. CD8+ T cells present low proliferation rate, diminished production of some Th1 cytokines, and reduced cytotoxic potential [70, 72–74]. Pleural effusion CD8+ T cells from lung cancer patients express cell markers associated with a memory phenotype (CD45RA-CD45RO+CD27+Granzyme AlowPerforin−), similar to those markers found in CD8+ T cells from chronic viral infections [24], which suggests that CD8+ T cells are exhausted.
Recently, pleural effusion CD8+ T cells, derived from lung cancer patients, have been shown to be susceptible to AICD. This phenomenon is preferentially observed in memory as well as terminally differentiated CD8+ T cells. AICD is associated with upregulation of FasL and TRAIL molecules. Interestingly, CD4+ T cells from malignant pleural effusions are not prone to AICD [75]. Thus, chronic stimulation by the lung tumor mass may sensitize CD8+ T cells to AICD, as it has been shown in TILs from various types of human cancers [75]. Nevertheless, evaluation of exhaustion in lung tumor-specific CD8+ T cells has not been possible, since lung tumor-associated antigens are not shared among all lung cancer patients [62].
Related posts:
 Role of Innate Immunity in Cancers and Antitumor Response
Role of Innate Immunity in Cancers and Antitumor Response
 Prognostic Value of Innate and Adaptive Immunity in Cancers
Prognostic Value of Innate and Adaptive Immunity in Cancers
 MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
 B Cells in Cancer Immunology: For or Against Cancer Growth?
B Cells in Cancer Immunology: For or Against Cancer Growth?
 Immunohistochemistry of Cancers
Immunohistochemistry of Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


