Fig. 4.1
Anti-CD40 mAb augmented antitumor responses of anti-CD3-activated TDLN cells via ligation to CD40 on both B cells and DCs. (a) Activated total unfractionated (Unfrac) TDLN cells were co-cultured with MCA 205 vs. MCA207 tumor cells to determine IFN-γ production. B cells were removed by CD19 depletion (CD19−), and DCs were removed by CD11c depletion (CD11c−). (b) Activated total TDLN (Unfrac) cells or B cell, DC-depleted TDLN cells (CD19− and/or CD11c−) TDLN cells adoptively transferred into tumor-bearing mice for therapy. *p < 0.05 compare with any other group in (a, b), respectively (Adapted by permission from the American Association of Immunologists, Inc. Copyright 2005: Li et al. [17])
In a separate study, Iuchi et al. reported that host B cells were required for adoptive transferred T cells to mediate optimal antitumor immunity [18]. Tumor-bearing mice were treated with adoptive transfer of T cells accompanied with IL-2 and IL-21 administration in wild-type and B cell knockout (B−/−) animals, respectively. They found that tumor growth inhibition was significantly diminished in the B cell-deficient mice after T cell + IL-2 + IL-21 combined therapy (Fig. 4.2).
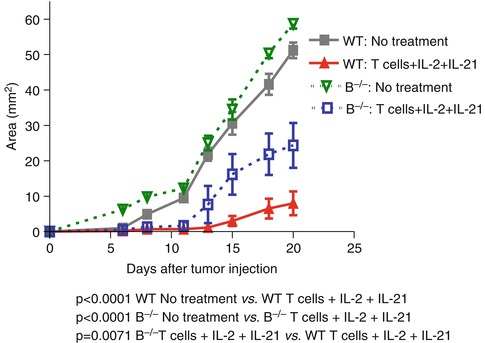
Fig. 4.2
Requirement for host B cells in T cell transfer + IL-2 and IL-21 administration-elicited antitumor immunity (Adapted by permission from the American Association for Cancer Research: Iuchi et al. [18])
In contrast to DCs, large numbers of B cells can be obtained from the blood of patients after ex vivo expansion (up to 1,000-fold) in the presence of CD40L [6]. For example, only about 106 DCs can be generated from 10 ml of blood, while 109–1010 B cells can be produced from the same volume of the blood sample. Additionally, CD40-B cells can be continuously expanded in long-term culture (>65 days) without the loss of APC functionality [6]. Therefore, CD40-B cells have the advantage over DCs in terms of isolation, generation, and long-term expansion. These characteristics make CD40-B cells a promising alternative as cell-based vaccines.
In current B cell vaccine preparations, activated B cells can be loaded with antigens by pulsing with peptides, proteins, tumor lysates, or by transfection with DNA or RNA, or transduction with viral vectors [9, 10, 19]. Coughlin et al. loaded RNA on CD40-B cells from pediatric patients. Vaccination using these B cells resulted in simultaneous targeting of multiple antigenic epitopes and induced CTLs [9]. Chung et al. reported that B cells stimulated with iNKT (CD1d-restricted invariant T cells) ligand alpha-galactosylceramide (alphaGalCer) could directly prime CTLs and generate long-lasting cytotoxic antitumor immunity in vivo [10]. Furthermore, Garbe et al. reported that semi-allogeneic fusions of microsatellite instability (MSI) tumor cells with B cells primed B cells to induce MSI-specific T cell responses [19].
4.3 Tumor Killer B Cells
B cells can directly kill tumor cells through antibody (Ab)-independent mechanisms [20]. Recent studies have shown that B cells express death-inducing ligands and can therefore mediate cell death under many circumstances. Evidence has emerged that B cells express Fas ligand (FasL), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), programmed death ligands 1 and 2 (PD-L1 and PD-L2), and granzyme B (GrB), which are potentially involved in B cell-mediated direct cytotoxicity against tumor cells [21–29].
Due to the well-known fact that B cells can produce Abs which lead to CDC and ADCC, as well as the recent findings that B cells may kill tumor cells directly through antibody-independent mechanisms, it is hypothesized that appropriately sensitized and activated B cells can function as effector cells to mediate antitumor immunity. Indeed, Li et al. [30] proved that in vivo sensitized and in vitro activated B cells could mediate tumor regression in cancer adoptive immunotherapy. In vivo sensitized TDLN cells were activated and expanded in vitro with LPS/anti-CD40, resulting in B cell proliferation and differentiation. These activated B cells were then adoptively transferred into tumor-bearing recipients for therapy. These tumor-primed and tumor-activated B cells significantly reduced lung metastases in an adoptive immunotherapy model (Fig. 4.3). Furthermore, total body irradiation (TBI) could enhance the antitumor activity of the adoptively transferred B cells. This study represents one of the early studies demonstrating that effector B cells could confer antitumor immunity after adoptive transfer into tumor-bearing mice [30].
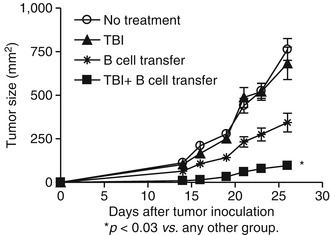
Fig. 4.3
TBI (total body irradiation) significantly augmented the therapeutic efficacy of adoptively transferred B cells in the s.c. D5 tumor model (Adapted by permission from the American Association of Immunologists, Inc. Copyright 2009: Li et al. [31])
Using a murine 4T1 pulmonary metastatic model, it was found that adoptive transfer of 4T1-primed and LPS-/anti-CD40-activated TDLN B cells significantly inhibited 4T1 pulmonary metastasis in tumor-bearing mice [31] (Fig. 4.4). The efficacy mediated by B cells was comparable to that mediated by an equal number of T cells, which served as a positive control in the experiment (Fig. 4.4a). Of note, adoptively transferred 4T1 TDLN T + B cells mediated inhibition of the spontaneous pulmonary metastasis of 4T1 in a dose-dependent manner (Fig. 4.4b).
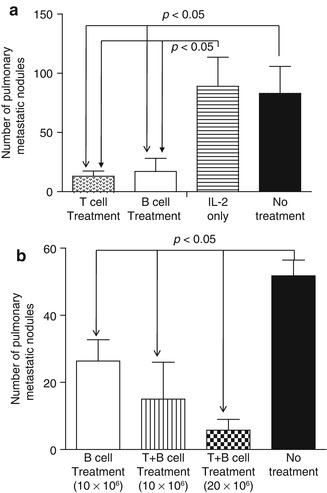
Fig. 4.4
(a) Adoptively transferred 4T1 TDLN B cells mediated effective inhibition of the spontaneous pulmonary metastasis of 4T1 breast cancer cells similarly to equal numbers of T cells. (b) adoptively transferred 4T1 TDLN T + B cells mediated inhibition of the spontaneous pulmonary metastasis of 4T1 better then B cells alone, and the efficacy was dose dependent (Adapted by permission from the American Association for Cancer Research: Li et al. [31])
This study also showed that activated 4T1 TDLN B cells caused tumor cell lysis directly in vitro in the absence of Ab and other effector cells and this direct cytotoxicity was tumor specific (Fig. 4.5). In these experiments, 4T1 mammary carcinoma murine tumor-primed TDLN B cells were activated with LPS and anti-CD40 mAb, washed thoroughly, and then co-cultured with 4T1 tumor cells or irrelevant tumor controls, Renca (renal cell carcinoma) and TSA (sarcoma). The effector B cells killed 4T1 cells directly in a dose-dependent way and were significantly more effective than their killing of the control tumors. These data support the conclusion that tumor antigen-primed and in vitro activated B cells are able to kill tumor cells independent of Ab or complement. However, the mechanism(s) by which the killer B cells lyse tumor cells directly in such a setting remains to be identified.
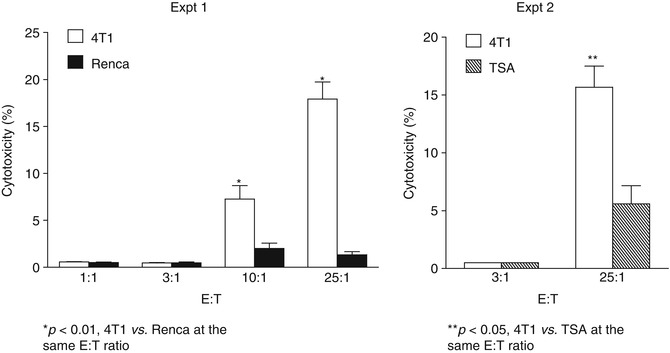
Fig. 4.5
Activated 4T1 TDLN B cells mediate direct and tumor-specific cytotoxicity of 4T1 cells (Adapted by permission from the American Association for Cancer Research: Li et al. [31])
In line with these findings, Kemp et al. demonstrated that CpG-A oligodeoxynucleotide (CpG-A ODN) stimulation of human PBMCs leads to high levels of functional TRAIL/Apo-2L expression on B cells, and these B cells mediate TRAIL-/Apo-2L-dependent tumor cell lysis [25].
Additional studies support the observation that B cell can function as effector cells in antitumor responses. For example, Penafuerte et al. reported that B effector cells activated with a chimeric protein consisting of IL-2 and the ectodomain of TGF-β receptor II (also known as FIST) induce potent antitumor immunity [32]. In this study, the B effector cells were characterized by the production of TNFα and IFN-γ and potent antigen presentation properties [32]. In addition, Forte et al. found that administration of a specific CD73 inhibitor, adenosine 5′-(α, β-methylene) diphosphate (APCP), to melanoma-bearing mice induced significant tumor regression [33]. They observed that after APCP administration, the presence of B cells in the melanoma tissue was more than that observed in control mice. This was associated with the production of IgG2b within the melanoma, implying a critical role for B cells in the antitumor activity of APCP [33]. Together, these studies suggest that the mechanisms underlining B cell-mediated antitumor immunity may involve multiple cellular and molecular events, as well as direct killing of the tumor cells.
4.4 Tumor-Infiltrating B Cells (TIL-Bs) in Cancer
Tumor-infiltrating B cells (TIL-Bs) have revealed controversial roles in antitumor immunity. They have been found in breast cancer [34–36], ovarian cancer [37], lung cancer [38], colorectal cancer [39, 40], cervical cancer [41], cutaneous melanoma [42], and prostate cancer (CaP) [43]. A few studies have indicated that TIL-Bs are correlated with favorable survival of patients [36, 37, 42, 44, 45], lower relapse rate [41], or low metastasis [42]. In a study on immune infiltrates in high-grade serous ovarian cancer, it was revealed that intraepithelial CD20+ TIL-Bs are associated with increased disease-specific survival [37]. Importantly, the association between the immune infiltrates and survival was dependent on histological subtype, because immune infiltrates were less prevalent in the other histological subtypes compared to the high-grade serous cases [37]. In breast cancer, TIL-Bs are present in about 24 % of tumors and comprise up to 40 % of the lymphocytic infiltrates [34]. TIL-Bs have been shown to undergo antigen-driven clonal proliferation and affinity maturation in situ [35]. Very recently, in a large patient cohort of different histological and biological subtypes, Mahmoud and colleagues provided evidence for a favorable outcome when high numbers of CD20+ TIL-Bs were present [36]. Additionally, TIL-Bs may be involved in humoral immune response in situ. Using recombinant Ab cloning techniques, Hansen et al. reported an antigen-driven humoral immune response directed against β-actin exposed on apoptotic mammary carcinoma cells [46]. Yasuda and coworkers identified TIL-Bs which produce tumor-specific Abs against mutated p53 [47]. Maletzki et al. also reported that TIL-Bs from colorectal carcinoma show an activated immunophenotype (CD23+, CD80+) and produce IgGs that specifically bind to allogeneic target tumor cells [40].
On the other hand, TIL-Bs may produce cytokines contributing to tumor development. It has been reported that TIL-Bs in castration-resistant CaP produce lymphotoxin by an inflammation-responsive IκB kinase (IKK)-β-dependent pathway, which then in turn activates IKK-α and STAT3 in tumor cells to enhance hormone-free tumor survival [43]. In this study, B cell infiltration was detected in 100 % of human CaP samples, while B cells were undetectable in normal prostate or benign prostatic hyperplasia [43]. Castration-resistant CaP growth was delayed in mice reconstituted with bone marrow from JH−/− mice, which lack mature B cells [43]. It was further found that these CaP allografts exhibited IKK-α nuclear translocation, which was dependent on IKK-β in B cells. IKK-β deletion abolished lymphotoxin expression by B cells. When lymphotoxin-β was ablated in B cells, growth of castration-resistant CaP was delayed. Similarly, another study showed that tumor-infiltrating T and B cells were not associated with long-term survival of patients with non-small-cell lung cancer [38].
The roles of TIL-Bs may be complicated, since the tumor environment is dynamic and changes during tumor onset and progression. TIL-Bs need to contact other immune cells or tumor cells to be activated or regulated, so their contributions to immune responses are likely to vary in different cancers and during the course of cancer.
4.5 Resting B Cells and Regulatory B Cells in Cancer
In contrast to activated B cells, there is abundant evidence indicating that resting B cells can promote the development or progression of cancer. Resting B cells are small B cells in the G0 stage of cell cycle, prior to activation. Studies have shown that B cell-deficient mice exhibit enhanced T cell antitumor activity and significant improvement in survival rate [48–52]. It has been reported that there are increased effector T cells [48], increased T cell infiltration of tumors [52], higher Th1 cytokine and antitumor CTL response [49, 51, 52], and even reduced T regulatory cell (Treg) frequencies [53] in these B cell-deficient mice. Some studies explored the possible mechanisms involved. B cells present in the priming phase result in disabled CD4+ T cell help for CTL-mediated tumor immunity [51]. B cells produce IL-10 which can repress antitumor immunity [49, 54]. Similarly, Abs were shown to promote primary tumor formation in a transgenic mouse model of inflammation-associated carcinogenesis [55]. Autoantibody responses to self-proteins triggered by cancer vaccines may influence the efficacy of vaccination [56]. Additionally, B cells have been shown to have other pro-tumorigenic roles. For example, enhanced NK cell antitumor activity has been reported in B cell-deficient mice [48, 50, 52]; however, the mechanisms are poorly understood.
We hypothesize that the effects of B cells on antitumor immunity depend on the presence of B cell subsets mainly involved under certain tumor conditions. In the past two decades, investigators have identified B cell subsets which are capable of suppressing the immune response. Suppression of an immune response was first reported in 1974 where spleen B cells were found to impair delayed-type hypersensitivity (DTH) responses in guinea pigs [57, 58]. This finding led to the conclusion that DTH responses and T cell function can be regulated by suppressor B cells. Subsequently, convincing data have demonstrated that IL-10-producing B cells, termed regulatory B cells (Bregs) by Mizoguchi et al. [59], can suppress inflammatory responses in experimental autoimmune encephalomyelitis (EAE), collagen-induced arthritis (CIA), and colitis [59–61]. Recently, Bregs and their potential immunomodulatory activities have been examined in several immune-related diseases. In the majority of these studies, the function of Bregs is dependent on IL-10 production, whereas the mechanisms are still undefined partly because of conflicting results regarding the phenotypic characterization of IL-10-producing cells. For example, the following B cells have been reported as putative mouse Bregs: CD1dhigh subset of B cells in chronic colitis in TCRα-deficient mice [59], CD21highCD23low B cells in contact hypersensitivity (CHS) mouse model [62], CD21highCD23high T2-MZ precursor B cells in CIA model [63], CD1dhighCD5+ B cells (termed B10 cells by Yanaba et al.) in CHS [64] and EAE models [65], CD138+CD19+ plasmablasts in Salmonella typhimurium infection [66], and T cell Ig domain and mucin domain protein (TIM)-1+ B cells [67]. For human, CD19+CD24hiCD38hi B cells have been found as putative Bregs [68, 69].
Triggering Toll-like receptors (TLRs) [70–72], the BCR [64], CD40 [73], or combinations thereof have been shown to promote IL-10 production by B cells. BCR-mediated Ca2+ flux appears to be required for IL-10 production, since B cells deficient in the calcium sensors stromal interaction molecule (STIM) 1 and STIM2 have a profound defect in IL-10 secretion and abrogated suppression abilities in vivo [74]. Nuclear factor of activated T cells (NFAT) 1, a transcription factor, is involved in Ca2+-dependent IL-10 production [74]. Therefore, their proposed model for IL-10 production by B cells is that, after BCR stimulation, STIM and Orai-dependent Ca2+ increase by store-operated Ca2+ entry (SOCE) activates calmodulin/calcineurin and then NFAT1, leading to IL-10 expression. In addition, the TLR signaling pathway is also required for IL-10 secretion [70–72]. Given that TLR stimulation does not induce Ca2+ mobilization in B cells, crosstalk between Ca2+ and Ca2+-independent TLR cascades may be involved in IL-10 production.
IL-10 is an immunomodulatory cytokine and inhibits Th1 polarization, prevents Th2 responses, and suppresses pro-inflammatory cytokine production by monocytes and macrophages [75]. So far, the potential role of Bregs in tumor immunology is not clear, but several studies suggest that Bregs can negatively regulate antitumor immunity. Using a mouse chemical carcinogenesis model, Schioppa et al. found that resistance to papilloma development in Tnf −/− mice was associated with a significant reduction in IL-10-producing B regulatory cells alongside an increase in IFN-γ-producing CD8+ T cells in the spleen [54]. In this study, Tnf −/− mice were resistant to chemical carcinogenesis of the skin. LPS-stimulated CD19+ B cells isolated from Tnf −/− mice produced less IL-10. These mice had a reduced absolute number of IL-10+CD19+ B cells in their spleens, and Tnf −/− mice were deficient for CD19+CD21high B cells. The authors speculated that resistance to carcinogenesis in Tnf −/− mice may result from increased CD8+ IFN-γ-producing T cells and decreased IL-10-producing B cells. In another study, Horikawa et al. reported that production of IL-10 by Breg inhibits lymphoma depletion during CD20 immunotherapy in mice [76]. They found that adoptive transfer of CD1dhighCD5+ B cells (that are enriched for B10 cells) eliminates the therapeutic benefit of CD20 mAbs in mouse lymphoma model. The transferred B10 cells in this model downregulated the expression of MHC II molecules and CD86 on macrophages and reduced LPS-induced nitric oxide and TNF-α production by macrophages, indicating that B10 cells suppress the antitumor response at least partly by downregulation of macrophage activity. Our unpublished data support that Bregs play a negative role in antitumor immunity. In melanoma and breast carcinoma models, depletion of IL-10-producing B cells from TDLN cells resulted in the generation of potent effector B cells which dramatically inhibit tumor metastasis after adoptive transfer in two genetically distinct immune competent hosts, B6 and Balb/c mice, respectively.
Although little is known about the mechanisms by which Bregs undermine effective antitumor immunity, several possibilities are suggested by studies on inflammation and autoimmunity. Bregs impair Th1 immune responses. The initial finding about Th1 response regulated by Bregs was reported by Skok et al. [77]; they found that IL-10 produced by B cells is involved in the feedback regulation of Th1 development. It has been reported that Bregs suppress the Th1 cell-mediated immune reactions in a number of mouse models, including EAE, CIA, CHS, and diabetes mellitus [60, 61, 64, 65, 72, 78, 79]. Fillatreau et al. reported that B cell IL-10 deficiency correlates with enhanced type I autoreactivity; in addition, transfer of IL-10+ B cells was found to result in resolution of EAE, characterized by enhanced encephalitogenic Th1 response [60]. Later, Lampropoulou et al. showed that TLR signaling in B cells suppresses inflammatory T cell responses (both Th1 and Th17) and stimulates recovery from EAE [72]. Similarly, using mouse model of CIA, Mauri et al. showed that transfer of IL-10-producing B cells inhibits T helper type 1 differentiation and prevents arthritis development [61]. Yanaba et al. also revealed that CD1dhighCD5+ B cell transfer normalized inflammation in CHS model [64]. Using NOD mouse model of type 1 diabetes (T1D), Hussain et al. found that BCR-stimulated B cells produce IL-10 and attenuate islet inflammation by polarizing CD4+ T cell response toward a Th2 phenotype [79].
Bregs induce the differentiation of Tregs. Given that μMT−/− B cell-deficient mice display reduced Treg frequencies in comparison with wild-type mice [53] and that these mice develop exacerbated EAE and Ag-induced arthritis (AIA) [60, 80], a role for Bregs in modulating Tregs was proposed. Several disease models have demonstrated that IL-10 produced by Bregs is important for the generation and/or maintenance of Tregs. Sun et al. reported that after oral tolerance induction, Treg cells increase much more in WT than in μMT−/− mice. However, adoptive transfer of B cells before treatment normalized Treg cell development in μMT−/− mice [81]. In this study, they found that sublingual tolerization with OVA/CTB (Ag conjugated to cholera toxin B subunit) enhances the tolerogenic activity of B cells and their production of IL-10, which was associated with the generation of Ag-specific Foxp3+CD25+CD4+ Tregs [81]. This relationship between Bregs and Tregs is further supported by the results from mouse models of airway sensitization. These results showed that Bregs prevent and reverse allergic airway inflammation via FoxP3+ T regulatory cells [82, 83]. Additionally, Bregs can induce the differentiation of T regulatory 1 (Tr1) cells [84–86]. Gray et al. reported that autoimmune inflammation could be protected by the induction of Bregs which induce T cell-derived IL-10 [84]. Blair et al. used the transitional 2 immature (T2) B cells stimulated with agonistic anti-CD40 (T2-like Bregs) to convert autologous effector T cells into Tr1 cells [86]. Sayi et al. also showed that B cells activated by Helicobacter TLR-2 ligands produce IL-10 and induce IL-10-producing CD4+CD25+ Tr1 cells depending on TCR signaling and a direct T-B cell interaction through CD40/CD40L and CD80/CD28 pathways [85].
4.6 Concluding Remarks
B cells are phenotypically and functionally heterogeneous. Characterization of B cell subpopulations is shown in Table 4.1. B cells play multiple roles in tumor immunity (Fig. 4.6). On one hand, accumulating literature indicate that B cells are significantly involved in antitumor responses. In this regard, B cells present tumor antigens to T cells to generate antitumor CTLs. Upon tumor antigen stimulation, B cells can differentiate into plasma cells to produce antibodies to target tumor cells via ADCC and/or CDC. In addition, B cells may act as killer cells to directly cause tumor cell lysis in the absence of antibodies. B cells migrate to tumor tissue and become TIL-Bs which may induce humoral immune response or act as killer cells in situ. On the other hand, regulatory B cells have been described which downregulate antitumor responses by producing immunomodulatory cytokine IL-10, suppressing Th1 immune responses, and enhancing Treg and Tr1 responses. Further characterization of B cell subsets responsible for these conflicting functions demonstrated in tumor immunity and understanding of the molecular mechanisms involved would help develop novel clinical strategies for cancer immunotherapy.
Table 4.1
Phenotypic characterization of B cell subpopulations
Marker | Source | References | ||
|---|---|---|---|---|
Resting B cells | Human | CD19+CD38−IgD+CD27− | Tonsils | |
CD38−IgM+IgD+CD27− | Blood | [88] | ||
Mouse | IgMlowIgDhighHSAlowCD21intCD23brightMel14 | Lymph node | [89] | |
(CD62L)brightCD44intCD69− | ||||
IgMhighIgDhighCD23bright | Spleen | [90] | ||
CD40 B cell | Human | CD19+CD38+CD80+CD86+CD71+ | Tonsils | [87] |
CD95+CPM(carboxypeptidase-M)+ | ||||
CD19+CD23+CD54+CD58+CD80+ | Blood | [6] | ||
CD86+MHCIhighMHCIIbright | ||||
Mouse | B7.1highB7.2highICAM+MHCIhigh | Spleen | ||
MHCIIbright | ||||
Putative Breg | Human
Related posts: Role of Innate Immunity in Cancers and Antitumor Response
Prognostic Value of Innate and Adaptive Immunity in Cancers
MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
Role of Plasmacytoid Dendritic Cells in Cancer
Envisioning the Application of Systems Biology in Cancer Immunology
Principles of Immunological Diagnostic Tests for Cancers Role of Innate Immunity in Cancers and Antitumor Response
Prognostic Value of Innate and Adaptive Immunity in Cancers
MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
Role of Plasmacytoid Dendritic Cells in Cancer
Envisioning the Application of Systems Biology in Cancer Immunology
Principles of Immunological Diagnostic Tests for Cancers
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||