patients with MDS.12 In a series of 915 patients with MDS referred to a tertiary care center, discordance in diagnosis using very strict criteria was documented in 12% of patients. A majority of patients were reclassified as having higher-risk disease by the International Prognostic Scoring System (IPSS).2 This has obvious implications, as most patients with higher-risk disease will be candidates for some form of therapy. Therefore, it is fundamental for the clinician to have proper documentation of the final morphologic diagnosis both for proper prediction of survival and for therapy selection.
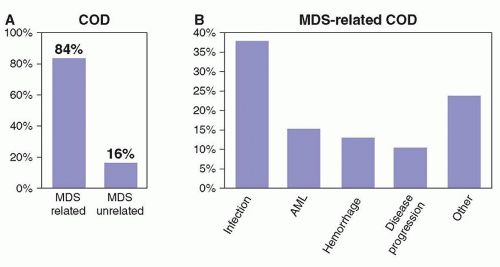 FIGURE 79.1. Cause of death in patients with myelodysplastic syndromes (MDS). A retrospective analysis was performed to determine the cause of death in patients with MDS. As shown in panel A, most patients succumb to causes intrinsic to the disease. B, The most common causes of death are infectious complications, transformation to AML, and bleeding. Adapted from Dayyani F, Conley AP, Strom SS, et al. Cause of death in patients with lowerrisk myelodysplastic syndrome. Cancer 2010;116:2174-2179. |
TABLE 79.1 WORLD HEALTH ORGANIZATION (WHO) CLASSIFICATION OF MYELODYSPLASTIC SYNDROME (MDS) | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a cytogenetic alteration will allow the differentiation of hypoplastic MDS from aplastic anemia.11 Finally, and more significantly, specific cytogenetic alterations have different prognostic values. A number of cytogenetic classifications have been proposed in patients with MDS. The most recent one is a 5-subgroup classification that forms the basis of the Revised International Prognostic Scoring System (IPSS-R).13,14 This cytogenetic scoring system is summarized in Figure 79.3. It is of interest as it further delineates rare cytogenetic alterations with favorable prognostic impact, such as alterations of chromosome 11, and also further defines the weight of alterations of chromosome 7 or complex karyotypes such as those with 3 or more abnormalities. Finally, the presence of specific cytogenetic alterations may help in the selection of specific forms of therapy. A classic example is the presence of deletions of chromosome 5 in patients with the so-called 5q-syndrome.15 Other alterations, such as chromosome 7 alterations or complex karyotypes may aid the clinician in selecting different forms of therapy with different intensities, such as hypomethylating agents.10 This is discussed later. A number of groups have proposed the use of fluorescence in situ hybridization (FISH) techniques to aid in the clinical work-up of patients with MDS.16 At the M.D. Anderson Cancer Center (MDACC), we do not routinely use FISH in MDS because it evaluates only a limited number of chromosomes, and the sensitivity and specificity of the different probes used is not fully understood or standardized. The frequency of cytogenetic alterations depends on risk. For instance, patients in the lower-risk categories by IPSS are diploid in over 50% of cases.17 This figure increases in patients with more advanced forms of the disease,18 and over 70% of patients with therapy-related MDS will have a cytogenetic alteration.19
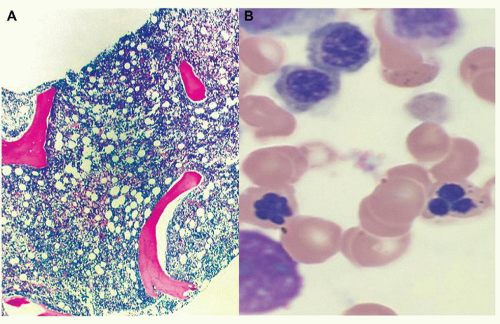 FIGURE 79.2. Representative example of morphologic alteration in MDS. Panel A shows a view of a bone marrow biopsy with almost complete replacement of marrow space with cellular element. Panel B shows a higher magnification of bone marrow aspirate, demonstrating nucleated red cell and micromegakaryocytes characteristic of this disease. |
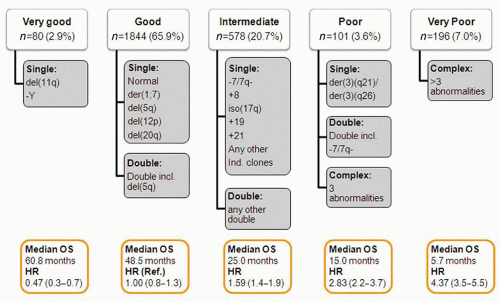 FIGURE 79.3. New 5-subgroup cytogenetic classification of myelodysplastic syndromes (MDS). This new cytogenetic classification divides patients into 5 categories based on their characteristics. Impact on survival is shown for each subset at the bottom of each subgroup. Adapted from Schanz J, et al.14 J Clin Oncol 2012;30:820-829. |
cancers. These include next-generation gene sequencing23 and analysis of single nucleotide polymorphisms.24 Although these assays are of great interest, they are not currently integrated into clinical practice.
of MDS.36 Of importance, several of these disorders are ribosomopathies characterized by altered ribosome biogenesis and function.37 Syndromes in this category include Diamond-Blackfan anemia, Schwachman-Diamond syndrome, dyskeratosis congenita, cartilage hair hypoplasia, and Treacher Collins syndrome.37 Haploinsufficiency in ribosomal genes, such as RPS14, are also implicated in the pathogenesis of the 5q-syndrome, thus providing further linkage between these conditions.38 Patients with Fanconi anemia are also at increased risk of developing MDS.39 Mutations in Runx1 have been described in MDS of patients with Fanconi anemia.40
TABLE 79.2 REPORTED FREQUENCY OF GENETIC LESIONS IN MDS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
fibrosis, number of marrow CD34+ cells, LDH, ferritin, and beta 2 microglobulin, to name a few. Prognostic stratification has an important role in MDS. One can consider this as a static concept helping the clinician predict survival and risk of transformation at the time of initial presentation without including the impact of any therapy. Other systems may allow prognostic calculation in a dynamic fashion by permitting sequential application during the life of the patient. And finally prognostic models may incorporate calculations of the impact of responses and or survival and response durations for a specific form of therapy. A number of classifications exist that fulfill one or more of these criteria. Until 2012, the standard prognostic classification system for patients with MDS was the IPSS.2 This model was developed in 1997 by Greenberg et al. and included a cohort of 880 patients that had not received prior therapy. This model has been the basis of most clinical research performed in the field over the last 20 years and therefore is of significant importance. Because IPSS is based on FAB morphologic criteria, in particular the percentage of marrow blasts up to 30%, and most currently approved drugs use either IPSS or FAB for their approval, IPSS still is of significant practical importance. IPSS is summarized in Table 79.3. IPSS has several limitations, the most important being that it underestimates the importance of the severity of cytopenias and it places too much weight on the percentage of blasts at the expense of cytogenetic alterations. Because of these limitations, a number of newer classifications have been developed by several groups. Examples include the WHO-based prognostic scoring system (WPSS) system63 and the Global MD Anderson Cancer (MDACC) model.18 That said, neither of these latter two models have been formally accepted
by all groups. Because of this, a very large international effort was initiated approximately 4 years ago to develop a new international MDS scoring system. This system is known as IPSS-R,13 or revised IPSS, and was recently published.13 IPSS-R is summarized in Table 79.4. The major differences between IPSS and IPSS-R is that the latter includes the new 5-subgroup cytogenetic classification discussed above and different cut-offs of cytopenias and percentages of marrow blasts, paralleled by 5 prognostic categories. IPSS-R has not been formally evaluated in a prospective fashion and has not yet been tested in patients receiving active therapy. Also, IPSS-R does not include molecular data and therefore is likely to be revised in the near future once large-scale mutational analyses are incorporated into routine clinical practice.
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


