Fig. 2.1
Fetal testis. a Fetal mouse: low magnification of a semi-thin section showing seminiferous tubules (ST) and interstitial tissue (I). b Fetal mouse: higher magnification of (a) showing gonocytes (G) in the central part of the sex cord with the Sertoli cells (S) around the periphery. The peritubular myoid cells (PMC) form a concentric layer around the cord, and Leydig cells (L) are present within the interstitium. c Fetal human: immunohistochemically labeled for the androgen receptor (AR) which is clearly expressed in PMC and in some interstitial cells. d Fetal human: immunohistochemically labeled for 3β-hydroxysteroid dehydrogenase (HSD3B) which is localized exclusively in the Leydig cells. In all photomicrographs, the bar represents 50 µm, Adapted from O’Shaughnessy and Fowler [22]. (c) Society for Reproduction and Fertility (2011). Reproduced with permission
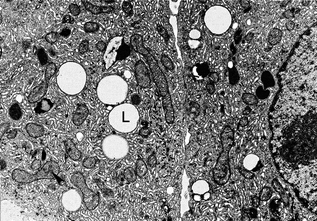
Fig. 2.2
Pubertal adult human Leydig cells. This electron micrograph of a resin section shows part of two adult human Leydig cells. Lipid droplets (L) can be seen along with numerous mitochondria (example is indicated with an arrowhead) and abundant smooth endoplasmic reticulum, Reproduced from Prince [200] with permission from the publisher John Wiley and Sons
In all mammalian species so far studied, two populations of Leydig cells have been identified. A fetal population that arises soon after testis differentiation, and an adult population which develops before puberty [3]. Until recently, it was thought that the fetal population regressed as the adult population developed, but evidence from the mouse suggests that the fetal cells remain present in the adult at about 10% of the total Leydig cell number [4]. In humans, blood levels of testosterone peak three times during development [5], and it has been suggested that this is evidence for three populations of Leydig cells in humans [6]. The first peak occurs at 12–14 weeks of gestation, during the fetal differentiation of Leydig cells [7, 8]. Testosterone levels then decline and are low for the remainder of gestation and the very early neonatal period. There is a second peak of testosterone, with associated high levels of INSL3, at 2 months postpartum that has been associated with the “extra” population of Leydig cells in the human (termed neonatal Leydig cells—see below) [9–11]. This post-natal testosterone surge is often referred to as “minipuberty” and may act to increase reproductive growth and alter neurobehavioral development in boys [12]. Beyond the neonatal period, Leydig cell numbers regress, although whether the fetal cells degenerate or remain in a morphologically unrecognized state is not clear. Either way, the interstitium contains few steroidogenically active cells during infancy [3]. The adult generation of Leydig cells starts to differentiate prepubertally but is not complete until adulthood [13] with serum levels of testosterone averaging 6 ng/ml (20 nmol/L) during adulthood [14]. Finally, there is a decline in testosterone secretion with aging, which is variable in its magnitude and time of onset. This age-related decline is multifactorial (see below), but it is likely to be associated with decreased testosterone production by the Leydig cells.
Fetal Leydig Cells
The testes begin to develop in the human fetus at around 6 weeks of gestation, with migration of the preSertoli cells from the coelomic epithelium and formation of the sex cords [15]. Fetal Leydig cells can be identified in the interstitial compartment by about eight weeks of gestation [16]. Studies in the mouse indicate that the fetal Leydig cells originate from two lineages: one arising from the coelomic epithelium and the other from cells associated with the vasculature along the gonad/mesonephros border [17]. Fetal Leydig cell numbers increase exponentially during the first half of the second trimester reaching a maximum number of about 2 × 106 around 18 weeks [7] before declining again toward birth [7, 18]. Testosterone is first detectable in the testis as early as 6–7 weeks of gestation [19] and rises toward the prenatal peak at 12–14 weeks [7, 20]. This peak in testosterone is due to the increasing number of fetal Leydig cells and to increasing levels of chorionic gonadotrophin (hCG) [21] which acts through the LHCGR to stimulate Leydig cell function. Testosterone levels decline in the second half of gestation as hCG levels drop markedly and Leydig cell numbers decline [8, 20]. Surviving Leydig cells in the second half of gestation are partly dependent on pituitary LH [22] for activity.
Neonatal Leydig Cells
Shortly after birth in the human, levels of LH rise and the number of Leydig cells increases leading to a neonatal surge in plasma testosterone levels at 2–3 months of age. At this stage, Leydig cells contain abundant smooth endoplasmic reticulum and mitochondria, as well as varying amounts of lipid droplets [13, 23, 24]. After the neonatal stage, Leydig cell numbers regress rapidly and become very scarce until six to eight years of age when they begin to increase toward adult levels. Well-differentiated Leydig cells are absent from the interstitial space during the quiescent phase, and in their place are partially differentiated Leydig cells and fibroblast-like cells. At this stage, Leydig cells are dispersed in a loose connective tissue matrix, and contain elongated nuclei with scarcely visible cytoplasm. It has been proposed that these partially differentiated Leydig cells and primitive fibroblasts are precursors of adult Leydig cells [25–27] although studies in the mouse would suggest other origins (see below). As suggested above, it has been proposed that the neonatal Leydig cells represent a third population of Leydig cells in the human, alongside the fetal and adult populations [6]. Given that fetal Leydig cells persist in the mouse, however, a simpler explanation would be that this neonatal peak of testosterone is due to re-activation of the fetal Leydig cells by the increase in LH levels which occurs at this time [4].
Adult Leydig Cells
The precursor cells for the adult Leydig cell population begin their transformation at 6–8 years of age, and the number of adult Leydig cells increases during puberty reaching a maximum reported as 8 × 108 per testis in the young adult [13]. At this time, a third rise in testosterone concentrations occurs, and levels remain high into middle/old age. Thereafter, Leydig cell numbers may decline in men as they age past 60 years, but there is current uncertainty about this as discussed below (see Leydig cell aging). Either way, between ages 20 and 60, there is a relatively stable equilibrium in the number of Leydig cells, which make up about 4% of the volume of the mature testis [28].
Steroidogenic Function of Leydig Cells
The main function of Leydig cells is to synthesize and secrete androgens, primarily testosterone. In humans, testosterone is synthesized mainly through the Δ5 pathway via dehydroepiandrosterone (DHEA) and androstenedione (Fig. 2.3) [29]. This requires the activities of four enzymes: cholesterol side chain cleavage (CYP11A1), 17α-hydroxylase/C17-20 lyase (CYP17A1), 3β-hydroxysteroid dehydrogenase/Δ5-4 isomerase (HSDB3), and 17β-hydroxysteroid dehydrogenase type 3 (HSD17B3) [30]. Leydig cells are the only cells in the testis which express CYP11A1, HSDB3, and CYP17A1 and are, therefore, the sole site for cholesterol conversion to C19 steroids. In contrast, data from the mouse show that the HSD17B3 enzyme is expressed only in the Sertoli cells of the fetal/neonatal testis [31, 32] which means that fetal Leydig cells primarily secrete androstenedione (the Δ4 pathway predominates in rodents) and Sertoli cells are required for testosterone synthesis. In the adult mouse, HSD17B3 is expressed solely in the Leydig cells and the Leydig cells alone produce testosterone [31, 32]. Whether this pattern of HSD17B3 expression and steroid synthesis is specific to the mouse or is also relevant to the human remains to be determined. During fetal development, testosterone from the fetal Leydig cells will masculinize the internal ducts and glands but masculinization of the external genitalia depends on the formation of DHT in the genital tubercle [33]. Formation of DHT can be from testosterone through the action of SRD5A2, as shown in Fig. 2.3, but an alternative pathway has also been described which bypasses testosterone, and this pathway may be equally important for fetal masculinization in the human [34].
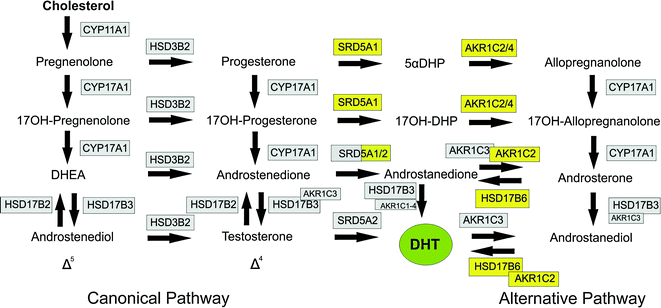
Fig. 2.3
Steroidogenic pathways leading to formation of dihydrotestosterone (DHT). The canonical pathways through Δ4 and Δ5 steroids are shown on the left with the alternative (backdoor) pathway on the right. Enzymes shaded in gray are required for the canonical pathway (with a number also required for the alternative pathway). Enzymes shaded in yellow are specific to the alternative pathway, Abbreviations; 5αDHP, dihyroprogesterone; 17OH-DHP, 17α-hydroxydihydroprogesterone; DHEA dehydroepiandrosterone; DHT, dihydrotestosterone, This Figure was published in Knobil and Neill’s Physiology of Reproduction, Vol 1, PJ O’Shaughnessy, Testicular Development, Pages 567–594, Copyright Elsevier (2015). Reprinted with permission
Androgen Synthesis via the Canonical Pathway
Androgen synthesis from cholesterol depends initially upon the transport of cholesterol from intracellular sources to the inner mitochondrial membrane, and subsequent loading of cholesterol into the catalytic site of CYP11A1 [35]. Cholesterol is hydrophobic and cannot cross the aqueous intermembrane space of mitochondria to reach the CYP11A1 enzyme rapidly enough by simple diffusion to support acute steroid synthesis [36]. To overcome this problem, the cholesterol is transferred into the mitochondria through the transduceosome which includes the steroid acute regulator (STAR) protein, translocator protein, and voltage-dependent anion channel [37]. In the inner mitochondrial membrane, cholesterol is converted by CYP11A1 to pregnenolone and isocaproaldehyde [38] which is unstable and quickly oxidized to isocaproic acid. This reaction requires a mitochondrial electron transfer system of adrenodoxin and adrenodoxin reductase to convey electrons from NAPDH to CYP11A1 [35].
Pregnenolone can act as substrate for testosterone synthesis through two different routes in the canonical pathway—the Δ4 and Δ5 pathways—so called because of the position of one of the double bonds on the steroid backbone. The particular pathway taken is species-dependent and is regulated by the relative affinities of the converting enzymes for different substrates. In humans, the pathway is predominantly Δ5 because CYP17A1 has a higher binding affinity for pregnenolone than HDS3B [39]. In rodents, CYP17A1 has a higher affinity for progesterone, and the Δ4 pathway predominates [40]. In human Leydig cells, therefore, the pathway is predominantly through pregnenolone, 17α-hydroxypregnenolone, DHEA, androstenedione, and testosterone.
The enzymes involved in the canonical pathway are located in the mitochondria (CYP11A1, HSD3B2) and in the smooth endoplasmic reticulum (CYP17A1, HSD17B3) [30, 41]. In humans, there are two genes encoding HSD3B (types I and II) and both enzymes show the same activity although only HSD3B2 is expressed in the Leydig cells [42, 43]. The CYP17A1 enzyme carries out two steps in the bioconversion of pregnenolone and progesterone to the C19 steroids, DHEA, and androstenedione, respectively, with 17α-hydroxypregnenolone or 17α-hydroxyprogesterone as transient intermediates, although human CYP17A1 fails to show detectable activity with 17α-hydroxyprogesterone as substrate [40]. The final step in the formation of testosterone is the conversion of androstenedione to testosterone, catalyzed by HSD17B3. This enzyme is part of a family of 14 enzymes that show related activities [44], and in humans, HSD17B1 and HSD17B5 (a member of the aldo/keto reductase superfamily, also known as AKR1C3) are also able to carry out the same reaction [44]. It is clear, however, that in the human fetal testis at least, the HSD17B3 is predominant since XY individuals lacking functional HSD17B3 activity have a significantly reduced ratio of testosterone/androstenedione and fail to masculinize externally during fetal development [45]. Interestingly, testosterone levels rise at puberty in these individuals and virilization does occur leading to the suggestion that HSD17B5 may be of importance in the adult Leydig cells [46].
Androgen Synthesis via the Alternative Pathway
Individuals who are genetic males but with disorders of activity in the enzymes of the canonical pathway will show disordered sex development (DSD) with incompletely masculinized external genitalia. Some cases of DSD (with normal androgen receptor signaling) cannot be explained by alterations in the canonical pathway, however, suggesting that other pathways/mechanisms are involved in androgen synthesis and masculinization [47]. In 2003, Wilson and colleagues described an alternative “backdoor” pathway of androgen synthesis in the testes of the pouch young tammar wallaby [48] (Fig. 2.3), and this pathway was subsequently shown to be active in the prepubertal mouse testis [49]. The importance of this pathway to human masculinization became apparent when unrelated individuals with DSD were shown to have disorders in one or more of the enzymes involved in the alternative pathway [34]. The non-functional/partially functional enzymes in these individuals were AKR1C4 and/or AKR1C2 which are both required for the alternative pathway but will not affect the canonical pathway (Fig. 2.3). Barring some other unknown mutation in these individuals, this would suggest that both canonical and alternative pathways to DHT synthesis are required during human male fetal development to ensure normal masculinization. Transcripts encoding enzymes involved in the alternative pathway have been shown to be present in the fetal human testis [34], and it has been assumed that testes secrete DHT via this pathway [34]. This is not clear, however, since earlier studies have reported little or no DHT synthesis by the human fetal testis [50, 51] suggesting that all DHT synthesis must take place at the target organ. It is also not clear whether the testes are the only organs involved in the alternative pathway since the fetal human liver also expresses at least some of the same enzymes [52]. Further studies are needed, therefore, to identify which tissues are involved in the alternative pathway, and to measure the levels of the different pathway intermediates in the fetal circulation.
Estrogen Synthesis by Human Leydig Cells
Estrogens are formed by aromatization of androstenedione or testosterone by the enzyme CYP19A1. In the testis of most species, aromatase activity is detectable in Leydig cells, Sertoli cells, and germ cells [53], and the relative contributions of each of these testicular cell types to testicular aromatase activity varies with age and between species. In the rat and mouse, Sertoli cells contribute to testicular aromatase activity in immature animals while germ cells are a significant site of activity in the adult [53]. Sertoli cells from juvenile Rhesus monkeys are reported to express aromatase activity, [54] although, in humans, Leydig cells appear to be the only source of testicular estrogens at all ages [55, 56].
Androgen Secretion by the Leydig Cells/Testis
Once synthesized, the lipophilic androgens move out of the Leydig cells by passive diffusion, down the concentration gradient. Within the testis, testosterone and precursors diffuse freely into the interstitial space and enter the testicular blood capillaries that are immediately adjacent to Leydig cells [57]. Interestingly, it is this process of testosterone release into the testicular vascular bed which might be altered in Klinefelter syndrome leading to reduced circulating testosterone levels [58]. Once they are part of the systemic circulation, secreted testosterone binds to plasma proteins and is present in both bound and unbound forms. In adult humans, more than 95% of testosterone is complexed with proteins, both the high affinity (K D = 1 nM) sex hormone binding globulin (SHBG) and the low affinity (K D = 1000 nM) albumin. The proportion of testosterone that is unbound or loosely bound represents the biologically active fraction, which freely diffuses from capillaries into cells. The SHBG-bound fraction is thought to act as a reservoir for the steroid, although SHBG-bound steroids may also enter cells via endocytic receptors on the surface of target cells and contribute to hormone action [58]. Increasing levels of SHBG during aging contributes to reduced free plasma testosterone during this period (see below, Leydig cell aging).
Other Functions of the Leydig Cell
The principal function of the Leydig cells is to produce androgen, but the cells are also the only source of INSL3 during fetal development [59, 60]. This hormone is essential, along with androgen, for inducing normal testicular descent although INSL3-receptors (RXFP2) are found on Leydig cells and on germ cells, and there is evidence that INSL3 can act to reduce germ cell apoptosis [61]. Any role that INSL3 plays in Leydig cell function, however, is likely to be restricted to the period around early puberty [61]. In order to identify other functions of the Leydig cells, Leydig cell-specific transcripts in the adult rat have been identified on the assumption that at least some of these transcripts are likely to be involved in cell-specific functions [62, 63]. Apart from transcripts encoding components of the steroidogenic apparatus, the most common predicted function of translated proteins from these cell-specific transcripts is endogenous and xeno-toxicant metabolism and reduction in oxidative stress [62]. The Leydig cells may, therefore, play a significant role in protecting the adult testis from damage caused by toxicants or by stress.
Regulation of Leydig Cell Development and Function
Leydig Cell Development
Most information available on the control of Leydig cell development comes from rodent models, and there is no reason to doubt that these fundamentals are significantly different in the human, but caution needs to be maintained when extrapolating between species.
Initial development of the fetal Leydig cells is dependent upon the Sertoli cells and, in particular, upon Desert Hedgehog (DHH) and Platelet-derived Growth Factor-α (PDGFA) released by the Sertoli cells [64–66]. In the absence of DHH in mice, there is a marked reduction in fetal Leydig cell numbers, reduced androgen levels and failure of masculinization, with a similar phenotype also seen in humans with a mutation in DHH [67, 68]. Similarly, reduced fetal Leydig cell numbers are seen in mice lacking PDGFA [69], and it is likely that the effect of both DHH and PDGFA is to promote expansion of the fetal Leydig cell precursor population [69]. In contrast, both NOTCH and WNT4 signaling act to inhibit fetal Leydig cell differentiation and WNT4 appears to be important in preventing Leydig cell development in the fetal ovary [70, 71]. Interestingly, downregulation of Wilm’s tumor gene (WT1) may also be required for fetal Leydig cell development, with over-expression in fetal Leydig cells leading to development of a Sertoli cell-like phenotype [72, 73]. A number of homeoproteins (ARX, LHX9, PBX1, and RHOX4) are expressed in interstitial cells, and they have been shown to be involved in testis development. Only ARX has, so far, been linked directly to fetal Leydig cell development [74], however, possibly through an action on the progenitor cells [75]. In rodents, secretion of LH by the fetal pituitary is not required for fetal Leydig cell activity [76, 77], and once formed, the cells appear to function largely independently of the fetal Sertoli cells [78]. Similarly, fetal Leydig cell activity in humans is not dependent on fetal pituitary LH [79] but, unlike rodents, activation of the LHCGR is essential [80], indicating that hCG is required in humans to stimulate Leydig cell activity in utero [22].
The adult Leydig cells develop from peritubular precursors [81–83] which may be stem cells [84]. After initial differentiation, the cells undergo one or two mitotic divisions to reach the final population size [85]. The initial process of adult Leydig cell differentiation is completely dependent on the Sertoli cells [78] since Sertoli cell ablation in the neonatal mouse leads to general failure of adult Leydig cell differentiation except in regions where Sertoli cells or Sertoli-like cells have survived ablation [78]. This failure of adult Leydig cell development is associated with apparent loss of Leydig cell precursor cells, although it is likely that the Sertoli cells are also directly involved in stimulating Leydig cell differentiation since DHH appears to be required for normal adult Leydig cell development, perhaps through inducing stem cell commitment [64, 86, 87]. The Sertoli cell-derived factor anti-Müllerian hormone (AMH) may also be involved in the regulation of adult Leydig cell development with a pubertal decrease in AMH required for normal maturation [88]. It has been shown that the orphan nuclear receptor NR2F2 is necessary for adult Leydig cell development, possibly through development of the progenitors and through maturation of the differentiated cells [89]. PDGFA is also involved in the differentiation process although the origin of this factor in the post-natal testis is not clear [90–93]. In addition to the requirement for Sertoli cells, it is clear that adult Leydig cell development is critically dependent on LH. Progenitor Leydig cells do not express the LH receptor [94], and initial Leydig cell differentiation is LH-independent [95, 96] but, in the absence of LH or the LH receptor, there is a marked reduction in the number of Leydig cells that develop in the adult [97]. Similarly, in mice with enhanced chronic exposure to LH activity, there is hyperplasia of the adult Leydig cell population [98]. Overall, therefore, the data suggest that initial Leydig cell differentiation is Sertoli cell-dependent/LH-independent, but that further development of the cells is critically dependent on LH.
One other factor of importance in Leydig cell development is androgen. In mice lacking androgen receptors, there is failure of normal adult Leydig cell development, with a reduction in Leydig cell number, and those cells which do develop lack many transcripts/proteins which are associated with the adult cell population [99, 100]. In mice with a more specific deletion of androgen receptors only in the Leydig cell, there is also inhibition of cell maturation although the effect is less marked than with complete androgen receptor deletion [101]. Leydig cell numbers are unaffected in Leydig cell-specific knockouts and so androgen effects on Leydig cell development appear to be a mix of direct effects on the cells and indirect effects through other cells which express androgen receptors such as the peritubular myoid cells and Sertoli cells. In humans lacking functional androgen receptors, Leydig cell dysfunction appears to be less marked than in the mouse, with circulating testosterone levels in the normal range, albeit with high circulating LH levels [102–105]. This difference may be accounted for by loss of CYP17A1 in mice lacking androgen receptors [106], as the CYP17A1 enzyme levels appear to be normal, or possibly increased, in androgen-insensitive humans [107]. Interestingly, in both mouse and human, loss of androgen receptors leads to late-onset Leydig cell apoptosis, an event that is very rare in normal Leydig cell populations [101]. Androgenic stimulation also appears to be required by the adult Leydig cell stem cells during fetal life to ensure normal stem cell numbers in the adult [108] which may explain evidence suggesting that reduced fetal androgen exposure is associated with lower adult male androgen levels [109].
Regulation of Leydig Cell Activity
The main regulator of Leydig cell activity is LH, and two types of responses to LH are seen. The first, acute response triggers a rapid production of steroid within minutes [110] through the binding of LH to the receptor, stimulation of adenylate cyclase, and the formation of the second messenger adenosine 3’,5’-cyclic monophosphate (cAMP) (Fig. 2.4). Increased cAMP causes subsequent phosphorylation of proteins via protein kinase A or C [111, 112] in a cascade of events leading to increased STAR phosphorylation and expression and increased testosterone synthesis. The increase in cAMP also leads to the activation of a Ca2+-signaling pathway [113] and increased NR4A1-mediated hormone-stimulated STAR expression [114]. In addition, LH-induced cAMP signaling in the Leydig cell transactivates the epidermal growth factor receptor (EGFR) leading to activation and expression of STAR protein [115] (Fig. 2.4). The second effect of LH is a long-term trophic effect on the Leydig cells mediated through transcriptional regulation. LH signaling acts through phosphorylation of transcription factors to enhance transcription from cyclic AMP-response elements (CREs) in the promoters for several LH target genes including STAR, CYP11A1, and HSD3B [110, 116]. Some target genes lack a consensus CRE, and expression is modulated by interaction between different transcription factors such as NR5A1 [117]. Overall, in the longer term, LH acts to maintain the steroidogenic enzyme activity of the Leydig cell and the steroidogenic apparatus. This can be seen clearly in animals which lack LH stimulation either through gene mutation/manipulation or after treatment such as hypophysectomy. In hpg or LHRKO mice, which both lack LH stimulation; the Leydig cells are smaller and contain large lipid droplets; steroidogenic enzyme transcript level/activity is markedly reduced and androgen output in response to trophic stimulation is very low [95–97, 118].
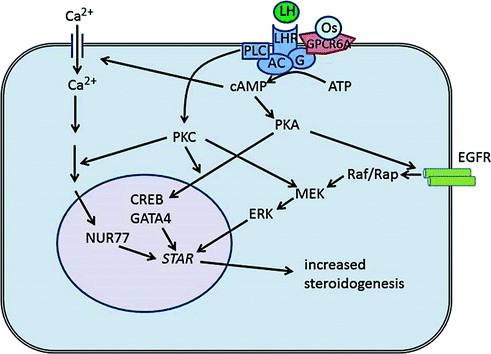
Fig. 2.4
Mechanisms of hormone-mediated steroid production in Leydig cells. LH is the major endocrine regulator of Leydig cell steroidogenesis, and binding of LH to the LH receptor (LHR also termed LHCGR) leads to activation of adenylate cyclase (AC). Osteocalcin (Os) also activates adenylate cyclase through binding to GPRC6A. Activation of adenylate cyclase leads to an increase in cAMP which, in turn, causes influx of Ca2+ into the cells and induces activation of various kinases including protein kinase A (PKA). This leads to activation of transcription factors (e.g. CREB, GATA4, NUR77) and induced expression of genes involved in steroid hormone synthesis, including STAR. Increased cAMP also activates the MAP kinase pathway via activation of the epidermal growth factors receptor (EGFR) which leads to increased expression and phosphorylation of STAR. The overall effect in the short term is to increase testosterone synthesis by the Leydig cells and, in the longer term, to increase transcription of the steroidogenic enzymes
In addition to LH, adrenocorticotrophic hormone (ACTH) can act to regulate fetal Leydig cell function in the mouse, although it does not appear to have any effect on adult Leydig cells [119]. Isolated fetal/neonatal mouse testes or Leydig cells will respond rapidly in vitro to physiological levels of ACTH with a marked rise in testosterone, similar to that seen in response to LH [119, 120]. It is not clear that ACTH plays a physiological role in development of the fetal Leydig cells in mice, however, since androgen levels are normal in mice lacking ACTH or ACTH and LH [121]. It remains to be seen whether ACTH is of importance to fetal Leydig cell function in species, such as the human, which require trophic endocrine support for normal fetal androgen production. Responsiveness of fetal Leydig cells to ACTH in the mouse does raise one interesting aspect of these cells and that is their similarity to adrenocortical cells. It has been reported that at least some of the fetal Leydig cells may derive from the same progenitor population as the adrenocortical cells [122], and at least some of the cells show distinct similarities both in hormone responsiveness and steroidogenic enzyme expression [123–125]. Whether these are a subpopulation of normal fetal Leydig cells or ectopic adrenal cells which have been shown to give rise to adrenal rest tumors in the testis [126] remains to be seen.
In rodents, the fetal Leydig cells are also responsive to a variety of local and endocrine factors such as pituitary adenylate cyclase-activating polypeptide, vasoactive intestinal peptide, and natriuretic peptides [127–129]. Since fetal Leydig cells in rodents do not specifically require LH, it is possible that activation of the cells in vivo is through multiple redundant mechanisms involving LH, ACTH, and activating peptides. A redundant mechanism such as this may have evolved to ensure that sufficient Leydig cell activation occurs to induce fetal masculinization. Whether human fetal Leydig cells are also sensitive to multiple agonists is not known, although it is clear that human fetal androgen production is critically dependent on LHCGR stimulation [80].
Recently, the bone protein osteocalcin has been reported to act as a trophic hormone on adult Leydig cells via the GPRC6A receptor [130, 131] (Fig. 2.4). Osteocalcin was shown to have a direct effect on testosterone synthesis by adult Leydig cells, and circulating testosterone levels and seminal vesicle weights were significantly reduced in mice lacking osteocalcin [130]. These effects were less marked than in mice lacking LH, but the fertility of the osteocalcin-deficient mice was reduced indicating that osteocalcin is required for optimal reproductive function. Evidence from two human patients with primary testicular failure linked to mutations in GPRC6A is also supportive of the hypothesis that osteocalcin can regulate testicular function [131]. The link between steroid hormones and bone mass has been known for a number of years but these data show that the skeleton can regulate Leydig cell function in a classic endocrine feedback loop [132].
In addition to the direct effects of LH, ACTH, and osteocalcin on Leydig cell function, follicle stimulating hormone (FSH) may play an indirect stimulatory role in the regulation of Leydig cells. Evidence for an effect of FSH comes from gonadotrophin-deficient mice and rats treated with FSH, and from differences in Leydig cell function between control mice and animals lacking FSH stimulation [133–137]. In addition to mice, there is also evidence that FSH can stimulate Leydig cell function indirectly in other species including humans [18, 138–140]. FSH receptors are restricted to the Sertoli cells [141], and so FSH effects on the Leydig cells must be mediated through Sertoli-secreted factors. The effects of FSH are fairly rapid, however, with a response seen in hpg mice in less than 4 h [135] suggesting that whatever paracrine factors are involved they must be acting directly on the Leydig cells. These effects of FSH are consistent with recent studies which show that ablation of the Sertoli cells in the adult mouse causes a marked reduction in Leydig cell number within 30 days [142], demonstrating that Sertoli cell factors are required for maintenance of the adult Leydig cell population. The identity of these factors remains unknown but an obvious candidate is DHH which is closely involved in Leydig cell development and continues to be secreted specifically by the Sertoli cells into adulthood.
Clinical Aspects
Leydig Cell Aging
Most studies show that plasma levels of total testosterone in men fall between 1% and 2% per year beginning at about age 40, although free testosterone declines more rapidly (~3% per year) as levels of SHBG increase at the same time [143–145] (Fig. 2.5). This decline in testosterone is multifactorial but can be divided into primary/compensated hypogonadism (low/normal testosterone with high LH) which is primarily linked to aging and secondary hypogonadism (low testosterone with low LH) which does not appear to be age-related but is clearly linked to obesity [146]. Metabolic clearance of testosterone slows with age [147] (which would tend to increase circulating testosterone), and so the increased LH seen in primary/compensated hypogonadism is evidence that the primary endocrine failure associated with aging is likely to be at the level of the testis [148]. Also, it has been shown that the circulating androgen response to increased LH declines with age in humans [148, 149] as might be expected from primary testicular failure.
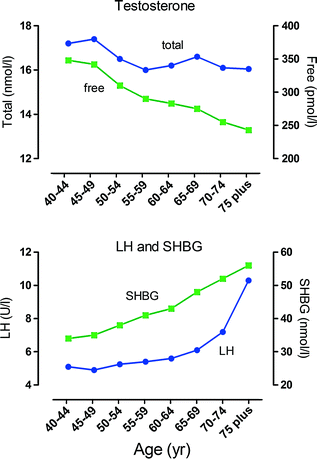
Fig. 2.5
Age-dependent changes in male hormone levels. These graphs show the relationship between age and hormone levels in men. Mean hormone values at 5-year age bands are shown based on data from 3220 men. Men with known pituitary or testicular diseases or current use of medications that could affect pituitary or testicular function or sex-steroid clearance were excluded. Total testosterone and free testosterone were significantly lower and luteinizing hormone (LH) and sex hormone binding globulin (SHBG) significantly higher in the older age groups. There was an apparent inflection point around 70 year for LH, Data shown in these figures are from [145] with permission of the authors
Age-related declines in testicular testosterone output could be caused by decreased Leydig cell numbers and/or reduced steroidogenic ability. There are a number of studies which report that Leydig cell numbers decline with age in the human population [150–153], although a more recent study found no age-related change in Leydig cell number [154]. Counting cell numbers in the testis is prone to technical problems which may have affected the older studies while the number of men over 60 in the more recent report [154] was only 4, so whether Leydig cell number declines with aging will remain uncertain until further studies are carried out.
Whether or not Leydig cell numbers decline with age, there is good evidence for degenerative changes in the cells including cytoplasmic or intranuclear crystalline inclusions, lipofuscin granules, diminished smooth endoplasmic reticulum, and smaller and fewer mitochondria when compared to young men [155–157]. Older men with higher serum LH and lower serum testosterone levels also have a large number of abnormal Leydig cells, suggesting that Leydig cell structural changes are related to changes in steroidogenic function [155]. Studies of intratesticular steroid levels in aging men do not show a specific lesion in the steroidogenic pathway but overall levels are lower, and there is evidence of reduced mitochondrial steroid production [158]. Using rat models in which the primary aging deficit is at the level of the Leydig cells, it has been shown that Leydig cell aging in the rat is associated with multiple defects in the steroidogenic machinery from reduced LH-dependent cAMP production to reduced steroidogenic enzyme levels [159]. This deterioration of Leydig cell function may be related to alterations in the redox balance of the cells leading to increased superoxide content with aging [159]. It is also likely, however, that other factors also contribute to the decline in Leydig cell function with age, as newly formed Leydig cells in testes from aged Norway rats show a rapid decline in steroidogenic function [160]. This does not appear to be due to changes in LH but may be caused by alterations in the levels of other trophic factors, changes in cell–cell signaling in the testis or vascular re-modeling associated with aging.
Leydig Cell Tumors
Leydig cell tumors were first identified by Sacchi in 1895, and are the most common interstitial tumors although they are rare overall and account for only 1–3% of testicular tumors [161–163]. Most Leydig cell tumors are unilateral (only 3% are bilateral [164]) and can appear at any age, although there is a peak incidence before puberty (between 5 and 10 years) and a second larger peak between 30 and 60 years. The tumors produce androgens, mainly testosterone, but serum estrogen levels may be elevated either through direct production of estrogen by the tumor or by peripheral aromatization of secreted androgen [165]. In boys, Leydig cell tumors are uniformly benign, hormonally active tumors, and account for about 10% of cases of precocious puberty [166]. In adults, most Leydig cell tumors are benign, and patients present with a painless testicular mass which may be palpable but is usually an incidental finding on scrotal ultrasonography for other conditions [167]. Small non-palpable Leydig cell tumors which are not visible on ultrasonograms can be seen by magnetic resonance imaging [168]. Where there is significant estrogen production, gynecomastia may be present along with loss of libido, erectile dysfunction, impotence, and infertility [165]. In adults, approximately 10% of Leydig cell tumors are malignant [162, 163] with regional lymph nodes, liver, lungs, and bone, the most common sites of metastases [162].
Macroscopically, the lesions associated with Leydig cell tumors are generally small, yellow to brown, well circumscribed and rarely hemorrhagic or necrotic. Microscopically, four different cell types can be found, ranging from large polygonal cells to spindle-shaped sarcomatoid cells [165, 169]. The cells have round nuclei with eosinophilic granular cytoplasm containing lipid vacuoles, lipofuscin granules, and Reinke’s crystals present in about one third of the cases [170] (Fig. 2.6). Ultrastructually, the cells show features expected of steroid-secreting cells, including abundant smooth endoplasmic reticulum.
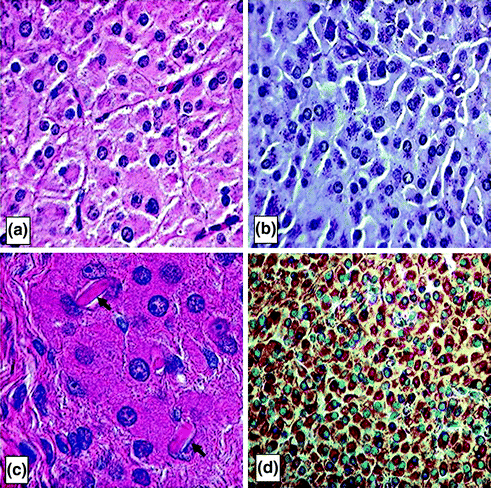
Fig. 2.6
Leydig cell tumors. Histologic sections of Leydig cell tumors. a Leydig cell tumor cells have abundant, eosinophilic cytoplasm with regular round nuclei, distinct cell borders and fibrovascular septa between the tumor cells. b Periodic acid Schiff stain which highlights the cytoplasmic lipofuscin granules in the tumor cells, a diagnostic clue to Leydig cell tumors. c Higher power photomicrograph showing intracytoplasmic Reinke crystals in a Leydig cell tumor (arrows). The crystals appear as refractile, cylindrical, rectangular, or rhomboid structures, and their identification is helpful for distinguishing Leydig cell tumors from other similar lesions (hematoxylin-eosin). d Leydig cell tumors typically show strong, diffuse cytoplasmic reactivity for α-inhibin (INHA) which is shown here as a brown stain. INHA is a sensitive and specific marker that can be used to separate testicular sex cord–stromal tumors, including Leydig cell tumors, from germ cell tumors, Reprinted from Al-Agha & Axiotis, An In-Depth Look at Leydig Cell Tumor of the Testis. Arch Pathol Lab Med. 2007; Vol 131 (issue 2): pp 311–317 with permission from Archives of Pathology & Laboratory Medicine. Copyright 2007 College of American Pathologists [165]
Classically, the primary treatment for Leydig cell tumors has been radical orchiectomy, and it remains in use for malignant cases. Testis-sparing surgery, with enucleation of the mass, has proved to be a feasible and safe choice, however, and is increasingly being reported for benign cases. A recent study of patients with Leydig cell tumors found a 100% disease-free survival with no local recurrences or metastases following testis-sparing surgery [171]. Testis-sparing surgery should also be considered for children who present with the clinical and biochemical findings typical of Leydig cell tumors, and an ultrasonographically defined encapsulated intratesticular mass [172]. Malignant Leydig cell tumors are radio-resistant and chemo-resistant, and have a poor prognosis with median survival time of 2 years [173].
The etiology of Leydig cell tumors remains uncertain, particularly in adults, but somatic activating mutations of the LHCGR have been found in a number of these tumors in boys [174]. This is consistent with the gonadotropin-independent nature of these tumors and with the development of fetal Leydig cell tumors in mice exposed to persistently high levels of hCG [175]. Other somatic mutations are also likely to be involved although a link to activating mutations in genes such as the Gs α-subunit of the stimulatory G protein (GNAS) have not been shown [174]. Leydig cell tumors in adults may be derived from the adult population of Leydig cells which would be consistent with the presence of Reinke’s crystals in some tumors as fetal Leydig cells lack these structures [176]. This may mean that the etiology of adult tumors differs from tumors in boys. The tyrosine kinase inhibitor imatinib has been reported to show chemotherapeutic activity in animal models [177], but this activity has not been demonstrated in an adult human trial [178].
Leydig cell hyperplasia shares the same clinical presentation as a Leydig cell tumor, including painful gynecomastia and decreased libido in adults, precocious puberty in children, and infertility or palpable testicular masses [179]. It should be noted, however, that many cases of apparent Leydig cell hyperplasia reported in the literature, in both humans and rodent models, are due to loss of germ cells (e.g., through cryptorchidism) causing shrinkage of the seminiferous tubules and an apparent increase in relative interstitial volume. Stereological measurement of Leydig cell numbers in these cases will often show no change in Leydig cell number per testis [180]. It is significant, therefore, that reported clinical Leydig cell hyperplasia is always associated with spermatogenic failure [179]. However, there is no doubt that true Leydig cell hyperplasia can occur if LH (or hCG) levels are elevated [179], and the hyperplastic Leydig cells are generally arranged in diffuse, multifocal, small nodules and show frequent mitoses, necrosis, and vascular invasion. Hyperplastic Leydig cells usually infiltrate between seminiferous tubules while benign Leydig cell tumors form nodules that compress surrounding tubules.
Leydig Cell Toxicology
As described above, the Leydig cells express a number of metabolic enzymes which might be expected to inactivate xenotoxicants and thereby protect the spermatogenic and steroidogenic function of the testis [62]. At the same time, however, the Leydig cells themselves are the potential target of a number of possible toxicants. This is an area which has seen a marked increase in publication activity in the last 10 years, with a considerable number of potential toxicants identified, and some of the better characterized/most relevant substances include phthalates, bisphenol A, statins, and ethanol. Phthalates are present in food packaging, cosmetics, and medical devices such as tubings and catheters, and so human exposure is significant with particular concern about fetal exposure [181]. Inhibitory effects of phthalates on rodent Leydig cell development and function are well documented, particularly with respect to the fetal Leydig cells, and may be related to altered expression of NR2F2 [182]. Changes to fetal Leydig cell development would be likely to affect normal masculinization of the fetus and are, therefore, potentially serious, but the effects of phthalates may be species-dependent with no clear effect seen in human fetal testis organ culture [181, 183], although there may be effects on adult human Leydig cells [184]. Bisphenol A is ubiquitous in the environment and is used primarily to manufacture polycarbonate plastic or as a non-polymer additive to other plastics and to epoxy resins. Bisphenol A has estrogenic activity and is classed as an endocrine disrupting compound, but it is also reported to directly inhibit fetal Leydig cell function in both rats and humans [185, 186] and is of ongoing concern. Statins act to inhibit cholesterol synthesis, and are taken daily by an estimated 20 million men, many of whom are more than 60 years old, and so with aging Leydig cells. Leydig cells need cholesterol as substrate for androgen synthesis (Fig. 2.3), some of which comes from circulating lipoproteins and some from de novo synthesis. A recent meta-analysis reported that statins reduce circulating testosterone concentration in men [187] which would be consistent with reported inhibitory effects in vitro on rat Leydig cells [188, 189], although no studies have yet been reported using isolated human Leydig cells. Long-term abuse of ethanol reduces circulating testosterone levels which may be a combination of direct effects on the Leydig cells [190, 191] with changes in circulating LH. Most studies report normal or elevated LH following alcohol abuse but the gonadotrophin response to pituitary stimulation is reduced suggesting altered hypothalamic pituitary function [190, 192, 193]. Finally, it has been shown that the alkylating agent ethane dimethane sulfonate (EDS) can act as a specific cytotoxicant in Leydig cells. This effect is species-specific with complete Leydig cell ablation seen in rats within 24 h but with little effect seen in the mouse, dog, and monkey [194]. It is still not known why the cytotoxic effect is specific to Leydig cells in the rat, but EDS has proven particularly useful in the study of Leydig cell biology [194].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


