Fig. 9.1
CD95: mRNA to protein
Once docked on CD95-DD, FADD self-associates [71] and binds procaspases-8 and procaspases-10, which are auto-processed and released in the cytosol as active caspases, which cleave many substrates leading to the execution of the apoptotic program and cell death. The complex CD95/FADD/caspase-8/caspase-10 is called DISC (Fig. 9.2) [10]. Due to the importance of DISC formation in the fate of cells, it is not surprising that numerous cellular and viral proteins were reported to hamper the formation of this structure, such as FLIP [72, 73] and PED/PEA-15 [74], which interfere with the recruitment of caspase-8/caspase-10 (Fig. 9.2).
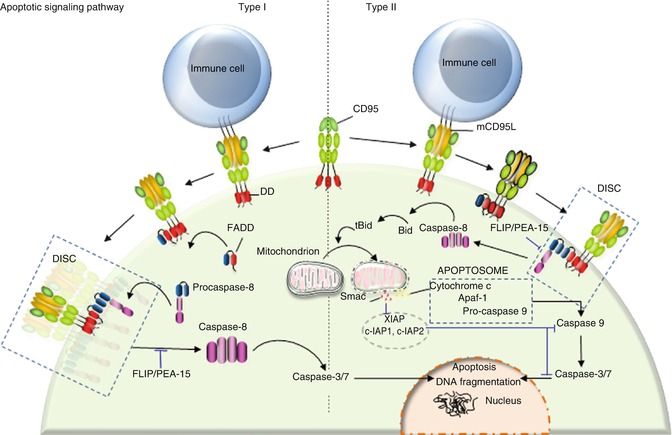
Fig. 9.2
Type I/II cells. Binding of transmembrane CD95L to CD95 leads to DISC formation. DISC consists of FADD and procaspase-8. C-FLIP and PEA-15 bind to FADD and prevent caspase-8 recruitment. At the DISC level, aggregation of procaspase-8 promotes its auto-cleavage and activation. Cleaved caspase-8 is then released in the cytosol where it promotes the cascade of caspase activation leading to apoptosis. Type I cells are characterized by an efficient DISC formation, which releases sufficient caspase-8 to directly activate caspase-3. By contrast, type II cells present a weak DISC formation, and the low amount of released caspase-8 activates the mitochondrion-dependent apoptotic pathway to amplify death signal
9.3.2 Type I/II Signaling Pathways
Following the discovery of CD95 and the first steps of its signaling pathway, Peter and colleagues described that cells can be divided in two groups with regard to the kinetics through which they respond to CD95-mediated apoptotic signals, the magnitude of DISC formation and the role played by the mitochondrion in this pathway [75]. DISC formation occurs rapidly and efficiently in type I cells releasing a large amount of activated caspase-8 in the cytosol, while type II cells have difficulty forming this complex, and the amount of active caspase-8 is insufficient to directly activate the effector caspase-3 and caspase-7 [75]. Nonetheless, type II cells experience cell death upon CD95 engagement and are even more sensitive to the CD95-mediated apoptotic signal compared to type I cells [75–77]. This discrepancy can be partly explained by the fact that the low amount of activated caspase-8 in type II cells is sufficient to cleave BID, a BH3-only protein, which constitutes the molecular link between caspase-8 activation and the apoptotic activity of mitochondria. Indeed, after cleavage by caspase-8, truncated BID (tBID) translocates to mitochondria, where it triggers the release of proapoptotic factors (Fig. 9.2) [78, 79]. Although CD95 stimulation activates the mitochondrion-dependent apoptotic signal in type I and type II cells, it seems that only type II cells are addicted to this signal as they display a higher amount of the caspase-3 inhibitor XIAP compared to type I cells [80]. Among the inhibitor of apoptosis protein (IAP) family, XIAP, c-IAP1, and c-IAP2 inhibit caspase-3, caspase-7 [81, 82], and procaspase-9 [83] activity by direct binding, thereby preventing access to substrates. Furthermore, XIAP can function as an E3 ligase whose activity is involved in the ubiquitination of active caspase-3 and its subsequent degradation through the proteasome [84]. To detach XIAP from caspase-3 and restore the apoptotic signal, cells require the release of SMAC/DIABLO (second mitochondria-derived activator of caspase/direct IAP-binding protein with low PI) by the mitochondrion [85, 86], explaining why type II cells are more addicted to this organelle compared to type I cells (Fig. 9.2).
To summarize, DISC formation and IAP amount are two cellular markers allowing a clear discrimination between type I and type II cells. Even though IAP overexpression can account for the mitochondrion dependency observed in type II cells, it remains unclear why DISC formation is hampered in type II cells and/or enhanced in their type I counterparts. Recently, high activity of the lipid kinase phosphoinositide 3-kinase (PI3K) or downregulation of its neutralizing phosphatase, phosphatase and tensin homologue on chromosome 10 (PTEN), was found in type II cells, while this signal is blocked in type I cell lines [87, 88]. The PI3K signaling pathway was reported to prevent the aggregation of CD95 [89], probably by retaining the receptor outside of lipid rafts [87, 90]. PEA-15, also known as PED, is a protein containing a death effector domain (DED) that has been shown to inhibit the CD95 and TNFR1 apoptotic signals (Fig. 9.2) [74]. Activation of PI3K and its downstream effector, serine-threonine kinase Akt, leads to phosphorylation of PEA-15 at serine 116 [87, 90]; this posttranslational modification promotes its interaction with FADD, ultimately inhibiting DISC formation [91, 92].
Notably, the existence of type I and type II cells is not only an in vitro observation, but has been identified physiologically in human body. CD95-mediated apoptotic signal cannot be altered in thymocytes or activated T cells expressing a Bcl-2 transgene, conferring to their type I nature [93], whereas hepatocytes expressing the same transgene resist CD95-induced apoptosis and thus behave as type II cells [94, 95].
9.3.3 What Can We Learn from CD95 Mutations?
Germinal mutations in APT–1 have been reported in patients developing a syndrome termed autoimmune lymphoproliferative syndrome type Ia (ALPS, also called Canale-Smith syndrome) [96–98]. ALPS patients show chronic lymphadenopathy and splenomegaly, expanded populations of double-negative α/β Τ lymphocytes (CD3+CD4−CD8−), and often develop autoimmunity [96, 97, 99, 100]. In agreement with the notion that CD95 behaves as a tumor suppressor, ALPS patients display an increased risk of Hodgkin and non-Hodgkin lymphoma [101]. Predominance of post-germinal center (GC) lymphomas in patients exhibiting either germ line or somatic CD95 mutations can be explained by the fact that, inside germinal centers of the secondary lymphoid follicles, the CD95 signal plays a pivotal role in the deletion of self-reactive maturating B lymphocytes [102], in addition to the fact that APT1 belongs to a set of rare genes (i.e., PIM1, c-myc, PAX5, RhoH/TTF, and Bcl-6) subject to somatic hypermutation [103, 104], which may affect biological function. In addition to post-GC lymphomas, significant amounts of mutations in the CD95 gene were found in tumors of various histological origins (reviewed in [54]). Extensive analysis of CD95 mutations and their distribution in APT–1 reveals that, with some exceptions, most are gathered in exons 8 and 9 encoding the CD95 intracellular region (Fig. 9.3) [105]. Remarkably, most of these mutations are heterozygous, mainly localized in CD95-DD, and lead to inhibition of the CD95-mediated apoptotic signal. Indeed, in agreement with the notion that CD95 is expressed at the plasma membrane as a pre-associated homotrimer [23, 24], formation of heterocomplexes containing wild-type and mutated CD95 prevents FADD recruitment and abrogates the ignition of the apoptotic signal in a dominant manner.
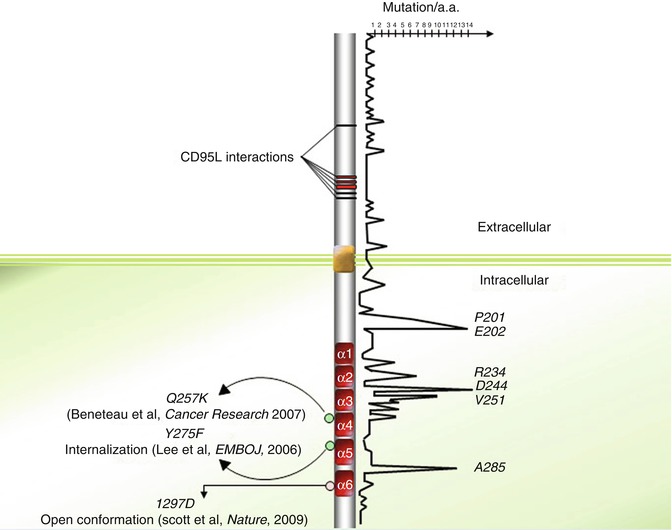
Fig. 9.3
Distribution of somatic and germinal mutations within CD95 protein sequence
Extensive analysis and positioning of various CD95 mutations described in the literature seem to highlight mutation “hot spots” in the CD95 sequence (Fig. 9.3). Among these hot spots, arginine 234, aspartic acid 244, and valine 251 account for a significant amount of the documented CD95 mutations. Indeed, among the 189 mutations annotated in the 335 amino acids of CD95, 30 (~16 %) are localized on these three amino acids (Fig. 9.3). Strikingly, the pivotal role played by these amino acids in stabilization or formation of intra- and inter-bridges between CD95 and FADD may explain these hot spots. For instance, both R234 and D244 contribute to the homotypic aggregation of the receptor and FADD recruitment [67]. Nevertheless, the observation of death domain hot spots is in contradiction with the study of Scott and colleagues demonstrating that the region of the CD95-DD interacting with the FADD-DD extends over a disperse surface through weak binding affinity [68].
Most ALPS type Ia patients affected by malignancies do not undergo loss of heterozygosity (LOH), which formed the hypothesize that preservation of a wild-type allele may contribute to carcinogenesis [106, 107]. In the same line, it was demonstrated that expression of a unique mutated CD95 allele blocks the induction of apoptotic signals, while it fails to prevent non-apoptotic signals such as NF-κB and MAPK [106, 107], whose induction promotes invasiveness in tumor cells [105, 108]. In addition, mutations found in the intracellular CD95-DD exhibit a higher penetrance of ALPS phenotype features in mutation-bearing relatives compared to extracellular mutations. These results suggest that unlike DD mutations, CD95 mutations localized outside the DD somehow prevent the apoptotic signal but may fail to promote non-apoptotic pathways, which may contribute to disease aggressiveness.
9.3.4 Regulation of the Initial Steps of CD95-Mediated Signaling
9.3.4.1 Lipid Rafts
In addition to CD95 downregulation or expression of the mutated allele of the receptor, the plasma membrane distribution of CD95 represents an additional pathway for tumor cells to develop resistance to CD95L-expressing immune cells. Indeed, the plasma membrane is a heterogeneous lipid bilayer comprising compacted or liquid-ordered domains, called microdomains, lipid rafts, or detergent-resistant microdomains (DRMs). These domains are described as floating in a more fluid or liquid-disordered 2-D lipid bilayer and are enriched in ceramides [109]. It has been elegantly shown that while CD95 is mostly excluded from lipid rafts in activated T lymphocytes, TCR-dependent reactivation of these cells leads to rapid distribution of the death receptor into lipid rafts [110]. This CD95 compartmentalization contributes to reducing the apoptotic threshold leading to the clonotypic elimination of activated T lymphocytes through activation of the CD95-mediated apoptotic signal [110]. Similarly, the reorganization of CD95 into DRMs can occur independent from ligand upon addition of certain chemotherapeutic drugs (e.g., rituximab [111], resveratrol [112, 113], edelfosine [87, 114, 115], aplidin [116], perifosine [115], cisplatin [117]). The molecular cascades that underlie this process remain elusive. Nevertheless, a growing body of evidence leads us to postulate that alteration of intracellular signaling pathway(s), such as the aforementioned PI3K signal [87, 90], may change biophysical properties of the plasma membrane, such as membrane fluidity, which in turn may facilitate CD95 clustering into large lipid raft-enriched platforms, favoring DISC formation and induction of the apoptotic program [118].
9.3.4.2 Posttranslational Modifications
Accumulation of CD95 mutations is not the only mechanism by which malignant cells inhibit the extrinsic signaling pathway. Posttranslational modifications in the intracellular tail of CD95, such as reversible oxidation or covalent attachment of a palmitic acid, were reported to alter the plasma membrane distribution of CD95 and thereby its subsequent signaling pathway. For instance, S-glutathionylation of mouse CD95 at cysteine 294 promotes clustering of CD95 and its distribution into lipid rafts [119]. This amino acid is conserved in the human CD95 sequence and corresponds to cysteine 304 (or C288 when subtraction of the 16 amino acid signal peptide is taken into consideration [12, 120]). Interestingly, Janssen-Heininger and colleagues emphasize that death receptor glutathionylation occurs downstream of caspase-8 and caspase-3 activation whose catalytic activity damages the thiol transferase glutaredoxin 1 (Grx1), an enzyme implicated in the denitrosylation of proteins [119]. The consequence of Grx1 inactivation is the accumulation of glutathionylated CD95, which clusters into lipid rafts, sensitizing cells to the CD95-mediated apoptotic signal. Based on these findings, caspase-8 activation occurs prior to aggregation of CD95 and redistribution into lipid rafts, both of which are requisite to form the DISC and subsequently activate larger amounts of caspase-8. In agreement with these observations, activation of caspase-8 was reported to occur in a two-step process. First, an immediate and small amount of activated caspase-8 (<1 %) is generated when CD95L interacts with CD95 that orchestrates acid sphingomyelinase (ASM) activation, ceramide production, and CD95 clustering, which in turn promote DISC formation and the outburst of caspase-8 processing essential to mount the apoptotic signal [121].
S-Glutathionylation consists in a bond between a reactive Cys-thiol and reduced glutathione (GSH), a tripeptide consisting of glycine, cysteine, and glutamate; its attachment to the protein will alter its structure and function in a manner similar to the addition of a phosphate [122]. S-Glutathionylation is not the only posttranslational modification of CD95 on a cysteine. S-nitrosylation of cysteine 199 (corresponding to C183 after subtraction of signal peptide sequence) and 304 (C288) in colon and breast tumor cells also promotes the redistribution of CD95 into DRMs, the formation of the DISC, and the transmission of the apoptotic signal [123].
Two reports have brought into light that covalent coupling of a 16-carbon fatty acid (palmitic acid) to cysteine 199 (C183) elicits the redistribution of CD95 into DRMs, the formation of SDS-stable CD95 microaggregates resistant to denaturing and reducing treatments, and internalization of the receptor [124, 125]. Although their order remains to be fine-tuned, these molecular steps play a critical role in the implementation of apoptotic signals.
Of note, similar to S-nitrosylation, both the aforementioned S-glutathionylation at C304 (C288) and palmitoylation at C199 (C183) promote the partition of CD95 into lipid rafts and enhance the subsequent apoptotic signal. Further investigation is required to address whether these posttranslational modifications are redundant and occur simultaneously in dying cells or are elicited in a cell-specific and/or in a microenvironment-specific manner. Understanding the molecular mechanisms controlling these posttranslational modifications would be of great interest in order to identify the mechanism by which tumor cells block them, leading to their resistance to the extrinsic signaling pathway.
9.3.4.3 CD95 Internalization
Using a powerful magnetic method to isolate receptor-containing endocytic vesicles, it has been shown that CD95 promptly associates with endosomal and lysosomal markers when incubated with an agonistic anti-CD95 mAb [126]. In addition, expression of a CD95 mutant in which the DD-located tyrosine 291 (Y275) is changed to phenylalanine does not seem to alter the capacity to bind FADD but compromises CD95L-mediated CD95 internalization occurring through an AP-2/clathrin-driven endocytic pathway [126]. More strikingly, expression of the internalization-defective CD95 mutant Y291F abrogates the transmission of apoptotic signals, but fails to alter the non-apoptotic signaling pathways (i.e., NFkB and ERK), and even promotes them (Fig. 9.3). These findings provide insight into the presence of a region in the DD, interacting with AP2 and promoting a clathrin-dependent endocytic pathway in a FADD-independent manner. Regarding the role of palmitoylation in receptor internalization, the interplay between lipid alteration and the AP2/clathrin-driven internalization of CD95 remains to be elucidated.
9.3.4.4 Ca2+ Response
It has been recently demonstrated that CD95 engagement evokes a rapid and transient Ca2+ signaling, which stimulates the recruitment of protein kinase C-β2 (PKC-β2) from the cytosol to the DISC [127]. This kinase transiently brakes DISC formation, providing a checkpoint before the irreversible commitment to cell death [128]. These findings raised the following questions: what are the Ca2+-dependent molecular mechanisms transiently inhibiting DISC formation, and do tumor cells use this signal to escape the immune response and/or resist chemotherapy?
9.3.5 Programmed Necrosis Also Known as Necroptosis
In 1998, inhibition of caspase activity was shown to sensitize fibroblastic L929 cell line to TNF-mediated necrotic cell death [42]. With respect to CD95 signal, Tschopp et al. showed that FADD and RIP1 participate in the implementation of a non-apoptotic signaling pathway, which leads to a necrotic morphology without chromatin condensation and with loss of plasma membrane integrity [41]. Of note, BID cleavage was not observed in this necrotic signal. While FADD plays a crucial role in both apoptotic and necrotic pathways, RIP1 recruitment to CD95 occurs independently of this adaptor protein. Indeed, yeast two-hybrid experiments showed that RIP1 can bind directly to the CD95 DD, while this interaction is lost when a bait corresponding to mutated CD95-DD (replacement of Val 238 to Asn) is used [129]. In addition, RIP3 (RIPK3, a member of the RIP kinase family) is an indispensable factor for the induction of the necrotic signaling pathway [78–80]. A growing body of evidence supports the existence of necroptosis (programmed necrosis). In addition, identification of necrostatin, a chemical inhibitor of necroptosis [130], which specifically inhibits RIP1 kinase activity [131], has accelerated the pace of discovery in this field of cell death. Interplays exist between apoptosis and necroptosis; for instance, caspase-8, a potent inhibitor of necroptosis for both CD95 and TNFR1 [132], plays a critical role in necroptosis by its ability to process and inactivate RIP1 and RIP3 [133, 134]. At least for TNF signaling, the necrotic signal relies on the activity of CYLD, a deubiquitinating enzyme that is also cleaved and inactivated by caspase-8 [135].
Overall, these findings suggest that the apoptotic machinery controls the necrotic one. This concept has been recently established in vivo by double-KO experiments [44–46, 136]. The KO of FADD or caspase-8 is deleterious in mice mainly by the fact that these two apoptotic factors are beneficial in inhibiting a RIP1-/RIP3-dependent necrotic signal; thus, their loss unleashes the necroptotic program and leads to embryonic lethality. Yet, most studies on necroptosis have focused on the TNF signaling pathway, whereas the mechanism by which CD95 can elicit this cell death pathway, and how the switch in this receptor occurs between non-apoptotic, apoptotic, and necroptotic signals remains unclear. Importantly, the impact of each cell death on antigen presentation, and on the efficiency of immune response after elimination of infected or transformed cells, remains unclear.
9.3.6 CD95L, an Inflammatory/Oncogenic Cytokine?
9.3.6.1 A Ligand to Create Immune Privileges
The transmembrane CD95L (CD178/FasL) is present at the surface of activated lymphocytes [64] and NK cells [137] where it orchestrates the elimination of transformed and infected cells. In addition, CD95L is expressed on the surface of neurons [138], corneal epithelia and endothelia [58, 139], and sertoli cells [59] to prevent the infiltration of immune cells and thus to prohibit the spread of inflammation in these sensitive organs (i.e., brain, eyes, and testis, respectively), commonly called “immune-privileged” sites. The description of physiological immune privilege was followed by tumor-mediated immune privilege, since two groups reported that the ectopic expression of CD95L by malignant cells participated in the elimination of infiltrating T lymphocytes and thus could play a role in the establishment of a tumor site whose access was denied to immune cells [140, 141]. However, these observations are controversial since ectopic expression of CD95L in allogenic transplant of β-islets [142, 143] and in tumor cell lines [144] led to a more rapid elimination of these cells than control cells, due to increased infiltration of neutrophils and macrophages endowed with antitumor activity.
9.3.6.2 At Least Two Different Ligands and Two Different Signals
Among the weapons at the disposal of immune cells, transmembrane CD95L contributes to the elimination of pre-tumor cells. Therefore, pre-tumor cells that escape the immunosurveillance will be shaped to develop resistance to CD95, a process termed immunoediting [145]. In other words, imprinting of the immune system on pre-tumor cells will select malignant cells with increased resistance towards the CD95L-induced signal. As previously mentioned, these alterations of the CD95 signal not only block the CD95-mediated apoptotic signal but also promote the transmission of non-apoptotic signals by CD95L, which may play a critical role in carcinogenesis [106–108, 146]. In agreement with this hypothesis, a complete loss of CD95 expression is rarely observed in malignant cells [147].
Accumulating evidence indicates that the apoptotic ligand CD95L behaves as a chemoattractant for neutrophils, macrophages [50, 143, 144], T lymphocytes [53], and malignant cells in which the CD95-mediated apoptotic signal is nonproductive [108, 148]. Nonetheless, the biological role of CD95L has to be clarified due to the fact that pathophysiologically the ligand is present in at least two forms with different stoichiometries. Indeed, CD95L is a transmembrane cytokine whose ectodomain can be cleaved by metalloproteases such as MMP3 [149], MMP7 [150], MMP9 [151], and ADAM-10 (A disintegrin and metalloproteinase 10) [152, 153] and released as a soluble ligand in the bloodstream. Based on the data demonstrating that a hexameric CD95L represents the minimal level of self-association required to signal apoptosis [154] and that cleavage by metalloproteases releases an homotrimeric ligand [154, 155], this soluble ligand has long been considered as an inert ligand competing with its membrane-bound counterpart for CD95 binding, thus acting as an antagonist of the death signal [155, 156]. It has been recently demonstrated that this metalloprotease-cleaved CD95L (cl-CD95L) actively participates in the aggravation of inflammation and autoimmunity in patients affected by systemic lupus erythematosus (SLE) by inducing the non-apoptotic NF-κB and PI3K [51, 53] signaling pathways (Fig. 9.4). Unlike transmembrane CD95L, induction of the PI3K signaling pathway by its metalloprotease-cleaved counterpart occurs through the formation of a complex devoid of FADD and caspase-8 which recruits the src kinase c-yes instead [53, 148]; this unconventional receptosome was designated motility-inducing signaling complex (MISC) [53, 157] (Fig. 9.4). Even though experiments by the authors did not detect any trace of caspase-8 in the MISC, this enzyme has been shown to participate in cell migration. The protease activity of caspase-8 can be abolished by its phosphorylation at tyrosine 380 by src kinase [158]. This posttranslational modification was observed in cells stimulated with EGF and in colon cancer cells exhibiting constitutive activation of src; from a molecular standpoint, this modification does not alter caspase homodimerization or recruitment in DISC [158]. Moreover, the EGFR-driven phosphorylation of caspase-8 at Y380 turns out to be a potent inducer of the PI3K signaling pathway by recruiting the PI3K adaptor p85 alpha subunit [159]. Ultimately, caspase-8 phosphorylation triggers cell migration. Nonetheless, it is noteworthy that CD95-induced migration and invasion do not appear to require an intact DD (reviewed in [160]), suggesting that either the caspase-8-dependent mode of cell migration occurs as an alternative signal for death receptors or that it only participates in non-death receptor-induced cell motility. It would be interesting to address this question in the future. To date, it can only be surmise that phosphorylation of caspase-8 at Y380 upon EGFR stimulation may prime certain cancer cells to become unresponsive to the apoptotic signal triggered by cytotoxic CD95L and meanwhile promote cell migration, an essential event in the course of cancer cell metastasis (Fig. 9.4).
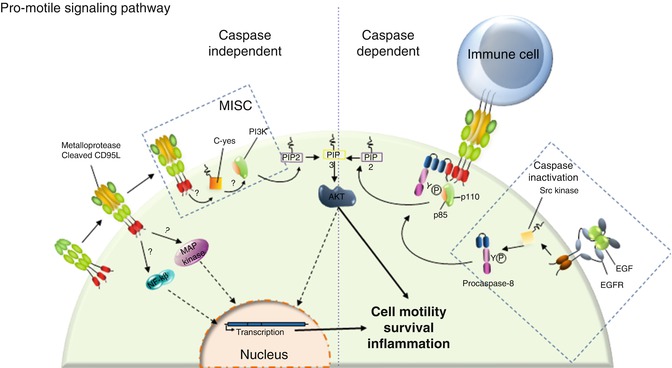
Fig. 9.4
CD95 triggers an unconventional PI3K signaling pathway. Left panel: In the presence of cl-CD95L, CD95 triggers MISC formation. This complex is devoid of FADD and caspase-8, but, instead, recruits the src kinase c-yes that implements the PI3K signaling pathway. CD95 engagement is also capable of NF-κB and MAPK activations through a yet unknown mechanism. Right Panel: It was reported that procaspase-8 can be phosphorylated by the tyrosine kinase src upon EGFR stimulation. This posttranslational modification not only blocks the catalytic activity of caspase-8 but also promotes the recruitment of the p85 subunit of PI3K. We surmise that this caspase-8 phosphorylation may favor the non-apoptotic signals induced by CD95
It is noteworthy that in a similar manner, a decrease in the plasma membrane level of CD95 or expression of a mutated CD95 allele, as observed in ALPS patients and malignant cells, inhibits the implementation of the apoptotic signal but does not affect the transmission of non-apoptotic signals, such as NF-κB, MAPK, and PI3K [106, 107, 147], suggesting that these signals may stem from a different domain than CD95-DD or rely on different thresholds to be elicited. In summary, although the CD95/CD95L interaction can eliminate malignant cells by implementation of the DISC or can promote carcinogenesis by sustaining inflammation and/or by inducing metastatic dissemination [50, 51, 53, 108, 147, 148, 161], the molecular mechanisms underlying the switch between these different signaling pathways remain enigmatic. An important question to be addressed is how the magnitude of CD95 aggregation controls the formation of “death”- vs. “motility”-ISCs. Addressing these questions will lead to the development of new therapeutic agents with the ability to contain the spread of inflammation or impede carcinogenesis at least in pathologies involving increased soluble CD95L such as cancers (e.g., pancreatic cancer [162], large granular lymphocytic leukemia, breast cancer [157], and NK cell lymphoma [163]) or autoimmune disorders (e.g., rheumatoid arthritis and osteoarthritis [164], graft-versus-host-disease (GVDH) [165, 166] or SLE [53, 167]). Altogether, these studies support the notion that the death function of CD95 may correspond to its “day job,” while the receptor may act as “a night killer” by fueling inflammation in certain pathophysiological contexts.
Strikingly, while the soluble form of CD95L generated by MMP7 (cleavage site inside the 113ELR115 sequence, Fig. 9.5) induces apoptosis [150], its counterpart processed between serine 126 and leucine 127 does not [51, 53, 155]. To explain this discrepancy, one may speculate that the different quaternary structures of the naturally processed CD95L underlie the implementation of a “death”- vs. “non-death”-inducing signaling complexes and downstream signals. In agreement with this notion, soluble CD95L bathed in the bronchoalveolar lavage (BALs) of patients suffering from acute respiratory distress syndrome (ARDS) undergoes oxidation at methionines 224 and 225 (Fig. 9.5), which enhances the aggregation level of the soluble ligand followed by its cytotoxic activity [168]. The same authors observed that the stalk region of CD95L, corresponding to amino acids 103–136 and encompassing the metalloprotease cleavage sites (Fig. 9.5), participates in the multimerization of CD95L, which accounts for the damage of the lung epithelium in ARDS [168]. Of note, in ARDS BALs, additional oxidation occurs at methionine 121 (Fig. 9.5), which in turn prevents the processing of CD95L by MMP7, and explains why this cytotoxic ligand keeps its stalk region [168]. Nonetheless, preservation of this region in soluble CD95L raises the question that whether an unidentified MMP7-independent cleavage site exists in the juxtamembrane region of CD95L, near the plasma membrane, or the ligand detected in ARDS patients corresponds to the full-length CD95L embedded in exosomes [169, 170]. Indeed, this peculiar exosome-bound CD95L can be expressed by human prostate cancer cells (i.e., LNCaP), and evokes apoptosis in activated T lymphocytes [171].
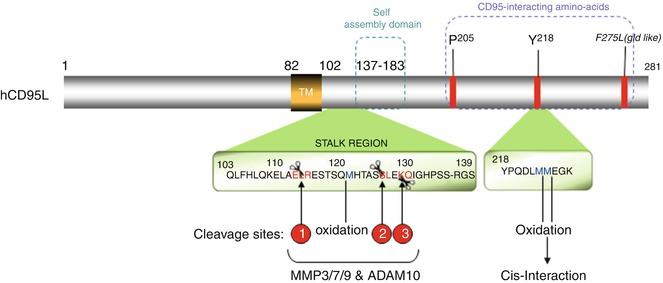
Fig. 9.5
CD95L: metalloprotease cleavage sites and domains
Overall, these findings emphasize that it will be of great interest in the future to finely characterize the quaternary structure of the naturally processed CD95L from the sera of patients affected by cancers or chronic/acute inflammatory disorders, to better understand the molecular mechanisms implemented by this ligand and thus predict its subsequent biological functions.
9.4 Concluding Remarks
Apoptosis is a fundamental process contributing to tissue homeostasis, immune response, and development. CD95, also called Fas, is a member of the tumor necrosis factor receptor (TNF-R) superfamily. Its ligand, CD95L, was initially detected at the plasma membrane of activated T lymphocytes and natural killer (NK) cells where it contributes to the elimination of transformed and infected cells. Given its implication in immune homeostasis and immune surveillance combined with the fact that various lineages of malignant cells exhibit loss-of-function mutations, CD95 was initially classified as a tumor suppressor gene. Nonetheless, in different pathophysiological contexts, this receptor is able to transmit non-apoptotic signals and promote inflammation and carcinogenesis. Although the different non-apoptotic signaling pathways (NF-κB, MAPK, and PI3K) triggered by CD95 are known, the initial molecular events leading to these signals, the mechanisms by which the receptor switches from an apoptotic function to an inflammatory role, and, more importantly, the biological functions of these signals remain elusive.
References
1.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57.PubMedCentralPubMed
2.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–90.PubMed
4.
Tatton WG. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Ann Neurol. 2003;53 Suppl 3:S61–70; discussion S70–2.PubMed
7.
Boldin MP, et al. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270(14):7795–8.PubMed
8.
Chinnaiyan AM, et al. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81(4):505–12.PubMed
Related posts:
 Role of Innate Immunity in Cancers and Antitumor Response
Role of Innate Immunity in Cancers and Antitumor Response
 Prognostic Value of Innate and Adaptive Immunity in Cancers
Prognostic Value of Innate and Adaptive Immunity in Cancers
 Nutrition, Immunity, and Cancers
Nutrition, Immunity, and Cancers
 B Cells in Cancer Immunology: For or Against Cancer Growth?
B Cells in Cancer Immunology: For or Against Cancer Growth?
 Immunohistochemistry of Cancers
Immunohistochemistry of Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


