Mastocytosis is a heterogeneous disease characterized by the abnormal growth and accumulation of mast cells in one or more organs. The first case of mastocytosis was reported in 1869 by Nettleship and Tay under the heading of “rare forms of urticaria.” Cutaneous lesions were described as “brown cutaneous lesions that wheal after scratching”1 and were termed urticaria pigmentosa (UP) 1 year after this initial description.2 The association between mast cells and mastocytosis was made in 1887, when Unna found that UP lesions were characterized by an increased number of mast cells in the dermis.3
Subsequent appreciation of the full clinical spectrum of these disorders has identified variants of mastocytosis that involve mast cell infiltration of visceral organs, and these forms of disease have collectively been termed systemic mastocytosis (SM).4 It is now accepted that SM may present with or without skin lesions and may show an indolent or aggressive clinical course, in some cases complicated by concomitant emergence of a clonal non-mast cell lineage disorder, such as one of the myeloproliferative disorders or myelodysplastic syndromes. This has led to further classification of mastocytosis based on hematologic findings, molecular markers, and serum levels of biomarkers like tryptase and the cluster designation (CD) markers such as CD25, thereby grouping patients into better-defined clinical categories, which have been adopted by the World Health Organization (WHO).5
PATHOPHYSIOLOGY
The current understanding of the etiology of mastocytosis has evolved from concentrated research efforts in the 1990s that linked two key factors controlling mast cell growth and activity—stem cell factor (SCF) and the SCF receptor, KIT, and disruption of the normal function of KIT through activating mutations.6
Mastocytosis is thus recognized in the majority of cases to represent a clonal disorder of a pluripotent hematopoietic progenitor cell with the most common mutation consisting of an activating mutation at codon 816 in KIT.6,7,8 and 9 This precursor is believed to be more primitive than precursors committed to either the neutrophil/macrophage or erythroid cell lineages, with the affected clone showing variable expansion in these lineages in the peripheral blood of patients with SM.10 Thus, mastocytosis in such cases represents a somatic cell disorder that is confined to hematopoietic lineages. Although T and B cells of such mastocytosis patients carry a codon 816 mutation (such as D816V), unlike mast cells, these cells do not express surface KIT when mature, and thus may be less susceptible to the biologic effects of constitutively activated KIT.
Early research efforts in the area of mast cell biology concentrated on identifying the key biochemical pathways and mediators that accounted for the clinical sequelae seen in physiologic processes due to mast cell activation, including anaphylaxis and allergic diseases. Subsequent work by Kitamura et al.11,12 focused on murine models of mast cell deficiency via studies on mice that exhibited abnormal Kit (W/Wv mice)11 and laid the foundation for suggesting the pathophysiologic basis of human mastocytosis as compromising dysfunction at the c-kit-SCF axis.12 Two predominant hypotheses thus emerged regarding the etiology of mastocytosis: (a) mastocytosis as a result of local overproduction of soluble SCF and (b) mastocytosis as a result of mutations in KIT and downstream signaling molecules that lead to cell proliferation. Over time, with the lack of evidence of overproduction of SCF in tissues13 and with the identification of the D816V mutation in KIT in patients with mastocytosis,6 the second hypothesis rose to prominence.
Human mast cells normally reside in tissues associated with epithelial surfaces, blood vessels, nerves, and glands, and are derived from CD34+ pluripotential progenitor cells.14,15 Except for a population of mast cells that reside in the bone marrow, mast cells complete maturation in peripheral tissues. During this maturation, mast cells downregulate CD34, but continue to express cell-surface CD117.14,15,16 Under normal physiologic conditions, mast cells and mast cell progenitors do not or only minimally express CD2, CD25, or CD35 on their cell surface. This expression pattern, however, is altered in most patients with mastocytosis.17
The KIT mutation D816V or another similar activating mutation in KIT is found in the majority of adults with SM. However, this single mutation cannot alone explain the multiple variants of mastocytosis. In one hypothesis, c-kit D816V would play an important and possibly causative role in indolent mastocytosis, where the pathologic hallmark is mast cell differentiation and clustering without signs of substantial proliferation. More advanced cases of mastocytosis would then require additional genetic defects not specific to mastocytosis; for example, a mutation in TET2 or NRAS18 that would contribute to the excessive proliferation of mast cell progenitors.
Human mast cells are heterogeneous in terms of morphologic, biochemical, and functional characteristics. These differences correlate with differences in their anatomic locations and, when perturbed in function, associate with specific clinical signs and symptoms. Clinical sequelae of mastocytosis are thus not only the result of organ infiltration, but also the result of mast cell mediator release from spontaneous or induced mechanisms.
Mast cells are deemed to be long-lived cells, though it appears that at least some mast cells may proliferate locally in tissues in response to inflammatory or repair processes. In tissue sections, mast cells typically appear as either round or elongated cells, usually with a nonsegmented nucleus with moderate condensation of nuclear chromatin, and contain prominent cytoplasmic granules and lipid bodies. The cytoplasmic granules of mast cells contain heparin and chondroitin sulfate proteoglycans covalently linked to a protein core. Under appropriate conditions, the proteoglycan complexes stain metachromatically with basic dyes.
Human mast cells are characterized primarily as either mucosal or connective tissue mast cells, the former being located at mucosal locations, such as the lamina propria of the gastrointestinal (GI) tract, and containing a specific tryptase (thus a “T-type” mast cell). The latter are more commonly found near the epithelial surface of the skin and respiratory, GI, and genitourinary tracts and containing both tryptase and a chymotryptase (CT mast cells).19 Human mast cell granules also contain such biologically active molecules as tumor necrosis factor-α, histamine, acid hydrolases, cathepsin G, and carboxypeptidase.20 They are activated by a number of stimuli that are both FcεRI dependent and FcεRI independent. After activation, mast cells immediately release granule-associated mediators and generate lipid-derived substances that induce immediate allergic responses. Together, these mediators are deemed responsible for many of the clinical sequelae of the immediate hypersensitivity reaction, including pruritus, flushing, palpitations, and lightheadedness, also commonly reported in patients with mastocytosis.
Major lipid mediators produced on appropriate activation via immunoglobulin E (IgE) or non-IgE stimuli include prostaglandin D2 and leukotriene C4.31 Mast cell activation is followed hours later by the synthesis and release of additional chemokines and cytokines, which then contribute to chronic inflammation. Growth factors and cytokines reported to be synthesized and released from mast cells include basic fibroblast growth factor, nerve growth factor, tumor necrosis factor, granulocyte-macrophage colony-stimulating factor, vascular endothelial growth factor, interleukin-1 (IL-1), IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-9, IL-10, IL-13, and IL-16.19,21
Studies performed in rodents, nonhuman primates, and humans have shown that many aspects of mast cell development are critically regulated by SCF, produced by such cells as endothelial cells, fibroblasts, and mast cells.19,21,22 SCF has been shown to work in concert with other growth factors, such as IL-3, IL-4, IL-5, IL-6, IL-9, and nerve growth factor, to induce optimal mast cell precursor proliferation and survival.14,15,20 SCF has also been reported to be present in the lesional skin of patients with UP23 but was not identified in blister fluids13 and has been reported within neoplastic mast cells.24 SCF augments mast cell mediator release in response to stimulation by IgE and antigen.22 Cells bearing mutated KIT on their surface have been shown to have an increased chemotactic response toward SCF.25 SCF may influence the aggregation of mast cells leading to mast cell lesions as found in SM within marrow.
The binding of SCF to KIT induces dimerization, with activation of intrinsic tyrosine kinase activity and phosphorylation of the receptor. This in turn leads to exposure of specific recognition motifs for intracellular binding proteins containing Src homology (SH2) domains such as phospholipase Cγ-1, phosphatidylinositol-3′ kinase, mitogen-activated kinase, and Ras protein.20,26 Activating or gain-of-function mutations in KIT are associated with constitutive tyrosine kinase activation and ligandindependent autophosphorylation of KIT, thereby giving affected mast cells a survival advantage over wild-type cells.20,27,28,29
The D816V mutation was first reported in patients with mastocytosis in the peripheral blood mononuclear cells (PBMCs) of adult patients with mastocytosis with an associated hematologic disorder and in patients with persistent mastocytosis and extensive disease,6,7,29 but codon 816 mutations (D816Y and D816H) have been shown to be present in UP lesions of adults and in a subset of pediatric patients with more severe mastocytosis.6,7,30 An E839K mutation has been reported in skin lesions of some pediatric patients with mastocytosis.31 Exceedingly rare KIT mutations, which are reported to be present in less than 1% of patients with mastocytosis, include R815K, D820G, V533D, V559A, del419, K509I, and A533D.32,33 KIT-dependent downstream signaling events including the JAK-STAT pathway may magnify the consequences of mutations in KIT.32 In addition to D817V, the V560G mutation within the juxtamembrane domain of KIT was detected in the human mast cell leukemia (MCL) cell line HMC-1,34 the E839K-dominant inactivating mutation in several reported cases of pediatric mastocytosis, and the rare germline mutation F522C.27,35 Thus far, somewhat different mutation patterns appear to be emerging for adult- versus pediatric-onset mastocytosis, which may explain the differences in presentation and clinical course for these two different patient populations.
Irrespective of effects on KIT, inhibition of mast cell apoptosis through other biologic pathways may also contribute to the pathogenesis of mastocytosis. A subset of SM patients with associated eosinophilia and increased serum tryptase levels has been described; these patients carry the Fip1-like-1-platelet-derived growth factor receptor-α (FIP1L1-PDGFRA) fusion oncogene in pluripotent hematopoietic progenitor cells, which results from an approximately 800-kb interstitial deletion of chromosome 4q12.36 Similarly, a rare case of SM and chronic basophilic leukemia was found secondary to a PRKG2-PDGFRB fusion.37 A polymorphism in the gene for the IL-4 receptor α-chain has been shown to be associated with less extensive mast cell involvement, with disease usually localized to the skin.38 In addition, the bone marrow cells of patients with mastocytosis have been found to constitutively express the antiapoptotic proteins Bcl-XL and Bcl-2,39 which may explain the long survival of these cells and perhaps their resistance to chemotherapy-induced apoptosis.
Additional studies using gene expression analysis of bone marrow mononuclear cells derived from patients with indolent SM revealed that, compared with healthy controls, patients with mastocytosis displayed a highly consistent profile with 168 genes that were significantly up- or downregulated in patient samples.40 Further analysis using such microarray technology will be useful to identify candidate genes distinguishing patient groups showing divergent clinical behavior.
Consistent with the progressive genetic instability that is often described in other human tumors, increased chromosomal abnormalities unrelated to the KIT locus have been detected in patients with more malignant forms of mastocytosis, and new abnormalities may appear in a subset of such patients on disease progression.41 No recurrent patterns of chromosomal changes have been consistently reported in patients with SM. The implications of these findings are that, at least in some patients with mastocytosis, the etiology of their disease may encompass a broader chromosomal problem of genetic instability. For instance, tumor mast cell lines express persistently high telomerase activity throughout the cell cycle which does not appear to be dependent on intracellular signals or cell replication, in contrast to normal human progenitor mast cells that experience transient induction of telomerase activity that is dependent on growth factor-mediated signals such as SCF-, IL-3-, and IL-6-mediated p38 mitogen-activated kinase and phosphatidylinositol-3′ kinase.42
CLINICAL FEATURES
The clinical manifestations of mastocytosis are diverse and may be divided into those that are systemic or localized.20,31,33 Systemic effects of this disorder result from the release of significant amounts of mast cell mediators into the circulation. Clinical signs and symptoms that comprise systemic mediator release are those reported with anaphylaxis and include flushing, pruritus, hypotension, syncope, palpitations, and tachycardia. GI symptoms are commonly associated with mastocytosis and include nausea, vomiting, abdominal cramping, bloating, and/or diarrhea. Peptic ulcer disease, which appears to reflect at least partially increased gastric acid secretion due to hyperhistaminemia, may occur in up to 50% of patients with systemic disease.43 Malabsorption, though less common, tends to be mild and may occur in those with progressive disease. Local sequelae of mastocytosis are largely due to the effects of mast cell collections at specific organ sites and may result in severe end-organ dysfunction due to infiltration of normal tissue with mast cells and subsequent fibrosis (e.g., end-stage liver disease due to fibrosis and bone marrow failure).
For some patients, in particular those with advanced disease or with an associated hematologic disorder, the most bothersome complaints include severe and nonspecific constitutional symptoms of fatigue, weakness, anorexia, weight loss, low-grade fevers, night sweats, musculoskeletal pain, headaches, depression, altered attention span, irritability, and even subtle cognitive deficits such as mild memory loss. Some of these symptoms are attributable to ongoing chronic disease, whereas others may in part be a result of the central nervous system effects of mast cell mediators.
Attacks in some individuals are precipitated by stimuli such as heat, cold, pressure, alcohol, medications (e.g., opiates, non-steroidal antiinflammatory agents, and estrogens), radiocontrast agents, and venoms. Reactions may be more severe in such patients (e.g., anaphylaxis after hymenoptera stings) because of an expanded mast cell population.21,44 Patients with aggressive disease also often present with lymphadenopathy, splenomegaly, or hepatomegaly that may or may not be symptomatic. One of the most difficult clinical scenarios of mastocytosis from a management perspective is the treatment of severe musculoskeletal pain and/or pathologic fractures due to the osteoporosis that results from release of mast cell mediators and/or an expanding marrow compartment with active proliferation of mast cells. Besides local disruption of normal bone architecture, mast cell infiltration of bone may cause bone loss due to the secretion of heparin, IL-6, proteases, and mediators, and from their paracrine effects on osteoclast function.
The most frequently involved organs in SM are the skin, bone marrow, lymph nodes, spleen, liver, and GI tract. The lungs are usually spared in mastocytosis. Atopy (e.g., eczema, allergic rhinitis) and airway hyperreactivity (e.g., asthma) are not generally features of this disease.
Mastocytosis is one of eight subcategories of myeloproliferative neoplasms in the 2008 WHO classification of tumors of hematopoietic and lymphoid tissues.5 The criteria for the diagnosis of cutaneous and SM adopted by the WHO are provided in Table 85.1. The diagnosis of SM requires that one major and one minor criterion or three minor criteria be present. WHO Criteria for variants of SM are provided in Table 85.2.
The prognosis of patients with adult mastocytosis is dependent on the extent of disease and presence of an associated hematologic disorder. Patients with ISM tend to remain within this category of disease, although a subset will progress to more aggressive forms of disease such as SM-AHNMD (associated non-mast cell lineage clonal hematologic disorder with systemic mastocytosis). For children with isolated UP, at least 50% of cases are reported to resolve by adulthood.45 Patients with SM-AHNMD have a course that depends largely on the prognosis of the specific hematologic disorder and response to aggressive therapy.20 The mean survival time for patients with MCL is usually less than 12 months, although the prognosis is improving with the use of newer tyrosine kinase inhibitors. The survival time with aggressive systemic mastocytosis (ASM) is 2 to 4 years with aggressive management. Variables strongly associated with poor survival in adult patients include advanced age, weight loss, anemia, thrombocytopenia, hypoalbuminemia, and excess bone marrow blasts.46
TABLE 85.1 WORLD HEALTH ORGANIZATION DIAGNOSTIC CRITERIA FOR CUTANEOUS AND SYSTEMIC MASTOCYTOSIS
Cutaneous mastocytosis (CM)
Typical clinical findings of urticaria pigmentosa (UP)/maculopapular cutaneous mastocytosis (MPCM), diffuse cutaneous mastocytosis (DCM), or solitary mastocytoma, and typical infiltrates of mast cells in a multifocal or diffuse pattern on skin biopsy.
Systemic mastocytosis (SM)
The diagnosis of SM is made if one major and one minor criterion are present, or if three minor criteria are met.
Major criterion
Multifocal, dense infiltrates of mast cells (15 or more in aggregates) detected in sections of bone marrow and/or another extracutaneous organ, and confirmed by tryptase immunohistochemistry or other special stains.
Minor criteria
In biopsy sections of bone marrow or other extracutaneous organs, more than 25% of the mast cells in the infiltrate are spindle-shaped or have atypical morphology, or, of all mast cells in bone marrow aspirate smears, more than 25% are immature or atypical mast cells.
Detection of an activating point mutation at codon 816 of KIT in bone marrow, blood, or another extracutaneous organ.
Mast cells in bone marrow, blood, or another extracutaneous organ express CD117 with CD2 and/or CD25.
Serum total tryptase persistently greater than 20 ng/ml in the absence of an associated clonal myeloid disorder.
WHO, World Health Organization.
TABLE 85.2 WORLD HEALTH ORGANIZATION CRITERIA FOR VARIANTS OF SYSTEMIC MASTOCYTOSIS
Indolent systemic mastocytosis (ISM)
Meets criteria for SM (see Table 88.1 ). No “C” findings (see below). No evidence of an associated non-mast cell lineage clonal hematologic disorder (AHNMD). In this variant, the mast cell burden is low and skin lesions are usually present.
Bone marrow mastocytosis
As above for ISM with bone marrow involvement, but no skin lesions.
Smoldering systemic mastocytosis
As above for ISM, but with two or more “B” findings and no “C” findings.
Meets criteria for SM and criteria for an associated, clonal hematologic non-mast cell lineage disorder, AHNMD (MDS, MPN, AML, lymphoma, or other hematologic neoplasm that meets the criteria for a distinct entity in the WHO classification).
Aggressive systemic mastocytosis (ASM)
Meets criteria for SM with one or more “C” findings. No evidence of mast cell leukemia. Usually without skin lesions.
Lymphadenopathic mastocytosis with eosinophilia
Progressive lymphadenopathy with peripheral blood eosinophilia, often with extensive bone involvement, and hepatosplenomegaly, but usually without skin lesions. Cases with rearrangement of PDGFRA are excluded.
Mast cell leukemia (MCL)
Meets criteria for SM. Bone marrow biopsy shows a diffuse infiltration by atypical, immature mast cells. Bone marrow aspirate smears show 20% or more mast cells. Mast cells account for 10% or more of peripheral white blood cells. Variant: leukemic mast cell leukemia as above, but less than 10% of white blood cells are mast cells. Usually without skin lesions.
Mast cell sarcoma (MCS)
Unifocal mast cell tumor. No evidence of SM. Destructive growth pattern. High-grade cytology.
Extracutaneous mastocytoma
Unifocal mast cell tumor. No evidence of SM. No skin lesions. Non-destructive growth pattern. Low-grade cytology
“B” findings
Bone marrow biopsy showing greater than 30% infiltration by mast cells (focal, dense aggregates) and/or serum total tryptase level greater than 200 ng/ml.
Signs of dysplasia or myeloproliferation in non-mast cell lineages, but insufficient criteria for definitive diagnosis of a hematopoietic neoplasm with normal or slightly abnormal blood counts.
Hepatomegaly without impairment of liver function, and/or palpable splenomegaly without hypersplenism, and/or lymphadenopathy.
“C” findings
Bone marrow dysfunction manifested by one or more cytopenia (ANC <1.0 × 109/L, Hb <10 g/dl, or platelets <100 × 109/L), but no obvious non-mast cell hematopoietic malignancy.
Palpable hepatomegaly with impairment of liver function, ascites, and/or portal hypertension.
Skeletal involvement with large osteolytic lesions and/or pathologic fractures.
Palpable splenomegaly with hypersplenism.
Malabsorption with weight loss due to GI mast cell infiltrates.
aWHO, World Health Organization.
Cutaneous Mastocytosis
Cutaneous mastocytosis is composed of three distinct clinical variants: UP/maculopapular cutaneous mastocytosis, solitary mastocytoma, and diffuse-erythrodermic disease, also known as diffuse cutaneous mastocytosis (DCM) (Table 85.1). UP is further subcategorized into four subvariants: Typical UP, a plaque form, a nodular form, and telangiectasia macularis eruptiva perstans (TMEP).5
The classic lesions of cutaneous mast cell disease are UP, which consist of reddish-brown macules, papules, or plaques that urticate (i.e., form a wheal and erythema with a distinct border when stroked [a positive Darier sign]). However, in a number of patients, cutaneous lesions are lacking, and other organs, particularly the bone marrow, when biopsied, support the diagnosis of SM.
UP lesions tend to occur in a generalized distribution, most commonly occurring over the trunk and generally sparing the face, scalp, palms, and soles. When abundant, they may form a cobblestone appearance. There appears to be no sex predilection or familial pattern of cutaneous disease, though mastocytosis of one form or another has been described in families, including several sets of twins.45
Histologically, UP lesions are composed of a collection of mast cells within the papillary dermis with variable extension throughout the reticular dermis and into the subcutaneous fat. An increase in dermal mast cells ≥ 10 times that of normal skin, in the absence of other pathology, is highly suggestive of UP.45,47 Petechiae, ecchymoses, and telangiectasias may be present in or adjacent to UP lesions. Blister formation and hemorrhage may occur, particularly in infants and young children. This complication is presumed to occur due to high local levels of mediators released from mast cells, but why this younger age group is more adversely affected is unknown. After age 10 years, vesicles do not generally occur, and UP lesions tend to be smaller and more numerous. Onset of UP lesions tends to follow a biphasic curve, with one peak at 2.5 months of age and another at 26.5 years.47 Of pediatric patients in whom UP occurs, approximately half lose these lesions by adolescence. The remaining patients generally have lighter macular lesions at previously involved sites. Pruritus is the most common symptom that accompanies UP. Approximately 15% to 30% of pediatric patients whose skin lesions persist into adulthood progress to develop SM.48
Although UP in adults may persist indefinitely, a subset of patients, estimated from 7% to 19% in published series, experience fading or resolution of cutaneous lesions over time.49 Regression of UP in patients with indolent SM (ISM) appears to parallel a decrease in disease severity in terms of constitutional symptoms, although bone marrow findings of ISM remain. In contrast, disappearance of lesions in patients with an associated hematologic disorder may herald progression of disease, with more severe bone marrow pathology noted on follow-up biopsies. The absence or presence of the Asp816Val mutation in PBMCs did not predict the course of UP. Disease progression in patients with SM-AHNMD is therefore better monitored with serum tryptase levels and bone marrow biopsy findings than with changes in the number, distribution, or intensity of skin lesions.
DCM is a less frequent cutaneous manifestation and generally presents before the age of 3 years. It is characterized by a diffuse mast cell infiltration of the dermis and thus the entire skin is generally involved. DCM presents as a yellow-red-brown discoloration with a peau d’orange appearance, or as a generalized erythroderma in which severe edema gives the skin a doughy appearance. Additionally, yellow-cream-colored papules have been described that resemble xanthomas and pseudoxanthoma elasticum.47 Only rarely does skin appear superficially normal in DCM. Dermatographism and formation of hemorrhagic blisters may occur. GI manifestations, such as diarrhea, flushing, and hypotension, may be associated, and such patients have an increased risk for more serious clinical sequelae such as shock, significant GI bleeding, and death. DCM may resolve spontaneously by age 5 to 15 months, but when persistent, the skin may remain thickened and doughy and recalcitrant to treatment. Solitary mastocytomas are considered a variant of cutaneous mastocytosis that may present at birth or more commonly within the first 3 months of age, with spontaneous involution during childhood.45,47 They are only rarely described in adults. They present as macules, plaques, or nodules and are formed by dermal collections of mast cells without cellular atypia. They are most commonly seen on the extremities and may involve the palms or soles. When systemic symptoms are present, they most commonly involve flushing.
TMEP is a rare form of cutaneous mastocytosis (<1% of cases) that is thought to usually be limited to the skin and only reported in adults. Select cases with concomitant splenomegaly, increased mast cells in the bone marrow, and abnormal skeletal radiographs suggest that this form of cutaneous disease may have systemic features.47,50 The characteristic skin lesion in TMEP is a telangiectatic, red macule on a tan-brown background. Individual lesions are 2 to 6 mm in diameter and are without sharply defined borders. Pruritus, purpura, and blister formation are not generally associated with TMEP, though lesions may become edematous when rubbed. Occasionally, these lesions are found to coexist with UP.
Indolent Systemic Mastocytosis
ISM is characterized by mast cell involvement at various organ sites, although significant organ dysfunction (C findings) is usually absent, and prognosis in these patients is generally good. In most cases, clonal mast cells express CD2 and/or CD25 surface markers and contain the D816V mutation. The bone marrow is the most common site of extracutaneous mastocytosis, with the vast majority of adult patients with indolent disease demonstrating bone marrow mast cell infiltration.51,52,53,54 The criteria for diagnosis of ISM are provided in Table 85.2.5
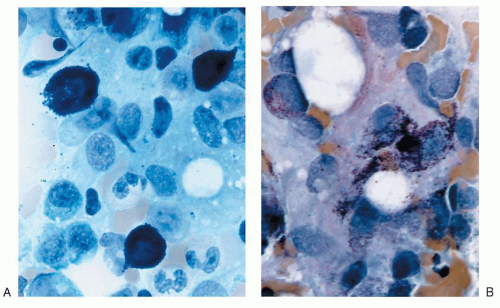
Characteristic bone marrow lesions of SM consist of focal dense, mixed, diffuse dense, or diffuse interstitial aggregates of mast cells, with differing histopathologic patterns of mast cell infiltration of the bone marrow related to differing prognoses and subtype of mastocytosis.6 Focal infiltrates are frequently observed in paratrabecular and perivascular areas and may be associated with adjacent lymphoid or eosinophil aggregates, or both.55 Mast cells in these patients show characteristic cytologic abnormalities, including cytoplasmic surface projections, eccentric oval nuclei, and a hypogranulated cytoplasm (atypical mast cell type I).5 A representative bone marrow aspirate from a patient with ISM with characteristic morphologic features compared with a control bone marrow aspirate is shown in Figure 85.1. Clonal mast cells generally express CD2 or CD25, or both, on their cell surface, and the D816V mutation.5,17,52 The bone marrow lesions of SM may contain a mixed population of B and T cells, in addition to mast cells identified by antitryptase staining. The histopathology of these lymphoid aggregates is similar to benign lymphoid aggregates associated with reactive bone marrow and not lymphoproliferative disease, the latter being uncommon in patients with adult-onset ISM. In bone marrow extensively involved by mast cell infiltration, the bony trabeculae may be moderately to markedly thickened.
FIGURE 85.1. Morphologic features of mast cells from normal versus mastocytosis bone marrow aspirate. Panel A shows staining of a control bone marrow aspirate from an individual with aplastic anemia using toluidine blue stain (magnification, 40×). Panel B shows a hematoxylin and eosin stain of a bone marrow aspirate acquired from a patient with indolent systemic mastocytosis and illustrates representative spindle-shaped mast cells with an eccentric nucleus (magnification, 40×).
A number of hematologic abnormalities have been reported in patients with SM, including cytopenias (e.g., anemia, thrombocytopenia, leukopenia, and lymphopenia) and increased blood cell counts (e.g., leukocytosis, eosinophilia, basophilia, monocytosis, lymphocytosis, and thrombocytosis).53 Hematologic abnormalities in children with mastocytosis are unusual, with a normochromic normocytic anemia being the most common finding.31 Prolonged bleeding times have been reported in infants with mastocytosis due to abnormal thrombin clotting times. In published reports of rare pediatric mastocytosis patients who have developed an associated hematologic malignancy, these were most frequently acute myeloid leukemias and acute lymphoblastic leukemias.31
Isolated bone marrow mastocytosis is a rare subvariant of ISM that is distinctive in lacking cutaneous and multiorgan involvement.5 The tryptase level in this group of patients is usually >20 ng/ml. These patients generally require no specific therapy, but ISM must be differentiated from ASM or MCL, in which skin lesions are also absent.
Only gold members can continue reading. Log In or Register to continue
 The Diagnostic and Therapeutic Approach to Hematologic Problems
The Diagnostic and Therapeutic Approach to Hematologic Problems



