Abstract
The estrogen receptor (ER) is the most important member of the nuclear receptor superfamily that controls the replication or apoptotic death of selected populations of breast cancer cells. The progesterone receptor is an estrogen-regulated protein in breast cancer that plays a role in predicting the hormone dependence of metastatic breast cancer. The mechanism of action of all effective antihormonal agents (tamoxifen, fulvestrant, aromatase inhibitors) is mediated through the ER signal transduction pathway and prevents estrogen action in the tumor cell. Tamoxifen is the pioneering selective ER modulator (SERM) that has estrogen-like effects in bone and the cardiovascular system, but antiestrogenic actions in breast cancer. The monthly depot injectable fulvestrant is a selective ER downregulator (SERD) that destroys the tumor ER and ERs throughout postmenopausal patient’s body. There is a current search for an orally active SERD.
Keywords
estrogen receptor, progesterone receptor, glucocorticoid receptor, androgen receptor, aromatase inhibitors, tamoxifen, fulvestrant, estrogen-induced apoptosis, selective estrogen receptor modulator, selective estrogen receptor downregulator.
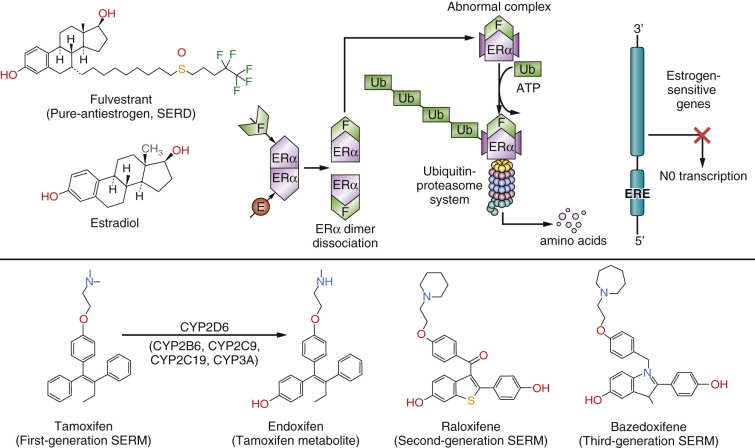
The nuclear steroid receptor (NSR) superfamily is essential for human life and reproduction. Similarly, the ability of breast cancer to subvert the positive role of NSRs in normal tissues to enhance survival becomes a vulnerability in the application of therapeutics. Not all members of the NSR superfamily are described in this chapter; the focus here is on the estrogen receptor (ER), the progesterone receptor (PR), the glucocorticoid receptor (GR), and the androgen receptor (AR) ( Fig. 21.1 ). However, it is the knowledge of the breast tumor ER that has had the greatest impact on the survival of patients, as the target that is recognized to be the most important in oncology and cancer therapeutics. The agents that bind to the ER and block estrogen-induced cell replication in breast tumors directly are selective ER modulators (SERMs): tamoxifen, raloxifene, and bazedoxifene ( Fig. 21.2 ). Another therapeutic agent that binds to the tumor ER, fulvestrant, is referred to as a “pure antiestrogen,” or a selective ER downregulator (SERD) (see Fig. 21.2 ). The agents that prevent estrogen synthesis in the peripheral tissues of postmenopausal women are aromatase inhibitors (AIs). The translational treatment strategy of long-term adjuvant antihormone therapy—to prevent estrogen action in the tumor—has extended or improved the lives of millions of women worldwide and continues to do so.
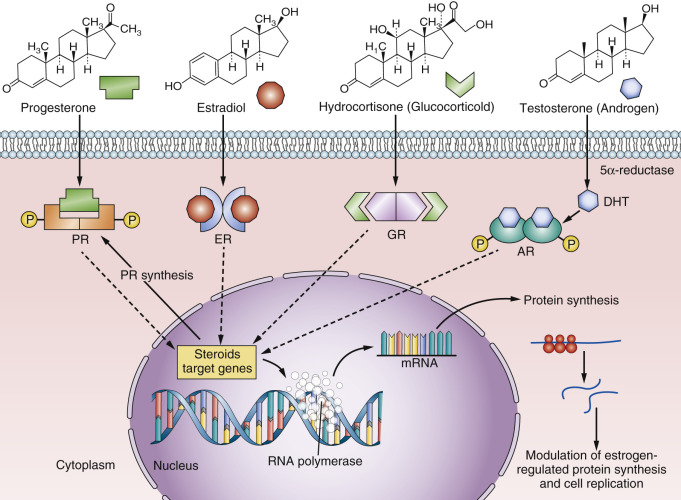
The PR has debatable value for refining the prediction of a successful outcome of adjuvant antihormone therapy over the ER alone for the treatment of breast cancer. Unlike the ER, which is the principal signal transduction pathway and the driver of breast cancer growth, the understanding of PR biology in breast cancer did not result in successful therapeutic advances to treat breast cancer. Nevertheless, the recent findings from the Women’s Health Initiative (WHI), in which estrogen-alone hormone replacement caused a paradoxical decrease in the incidence of breast cancer, whereas the combination of estrogen plus medroxyprogesterone acetate (MPA) causes an increased incidence in breast cancer, must be considered. This mandates a reevaluation of our basic understanding of the mechanism of action and interactions of estrogen and progestin in breast cancer tissue.
It is now clear that estrogen exhibits cytotoxic effects on estrogen-deprived breast cancer cells after menopause. This is placed into clinical context in this chapter, and the molecular modulation of estrogen action by glucocorticoids and androgens is considered. The AR is present in breast cancer. Although no therapeutic agents for breast cancer treatment have yet emerged as the standard of care, the AR is being considered a putative target in triple-negative breast cancer.
Estrogen Receptor
Historical Perspective
The evolution of our understanding of the hormonal control of breast cancer growth has recently been described in detail. Nevertheless, it is important to identify the landmark clinical observations and the discovery of the ER as milestones in translational medicine. These observations in the 20th century aided patients’ care despite the lack of a clear understanding of the molecular mechanism of estrogen action as the driver of breast cancer growth. Important new biological principles have now been deciphered during the 21st century that provide biological transparency and clinical rules for the care of patients as new standards of health care.
An evaluation of the first successful treatment for breast cancer was conducted by Stanley Boyd in 1900. He accumulated all of the clinical case reports of oophorectomy in premenopausal breast cancer patients to establish a response rate. He noted a 30% response rate to oophorectomy, which has stood the test of time and is noted with all other endocrine ablative, antiestrogenic, or high-dose estrogen therapies. The connection between estrogen and mammary cancer growth in laboratory animals correlated well with ovarian ablation in premenopausal breast cancer patients. Then a paradox occurred. Haddow and coworkers reported that high-dose synthetic estrogen administered to postmenopausal women with metastatic breast cancer (MBC), also had a 30% response rate; but only in patients that were treated more than 5 years past the menopause. This was a completely counterintuitive clinical observation, but despite the fact that there was no understanding of mechanisms, high-dose estrogen therapy became the standard of care for postmenopausal MBC until the discovery and clinical evaluation of tamoxifen in the mid-1970s. Tamoxifen, a nonsteroidal antiestrogen, blocks tumor growth by preventing estrogen binding to the ER. This antiestrogenic treatment strategy reaffirmed the principal role of estrogen to stimulate breast tumor growth.
It is important to state that only a minority of MBC patients during the 1960s and the early 1970s (before tamoxifen) responded to either endocrine ablation (oophorectomy, adrenalectomy, and hypophysectomy) or high-dose estrogen therapy. Responses were transient for a year or two. A predictive test was required for clinicians to decide which patients, who were the majority with MBC, would not respond to endocrine treatment. The goal was to reduce hospitalization and morbidity for those patients who would not respond. The discovery of the ER in estrogen target tissues (uterus, vagina, and pituitary gland) in animals and in some breast tumors in women created the first understanding of a potential molecular mechanism of estrogen action.
The hypothesis to be tested was straightforward; if there was no ER in the tumor biopsy, then no response would be expected to endocrine ablation therapy. Breast tumor growth would not be dependent on estrogen. The National Cancer Institute convened an international meeting in 1974 to review all of the clinical correlations for ER tumor content in biopsies in MBC, with the response of MBC to endocrine additive or ablative therapy (the final conclusions are illustrated in Table 21.1 ). What was originally a 30% response rate for unselected patients now segregated into a less than 10% response rate for patients with an ER-negative tumor biopsy, whereas patients with an ER-positive tumor biopsy had more than a 55% to 60% response rate. This conclusion from the meeting resulted in the introduction of the ER assay being required to determine the hormone-responsive status of all women with a diagnosis of breast cancer in the United States. At the time, this was a landmark advance in translational research to aid women’s health. However, the strategic targeting of tamoxifen, an antiestrogenic medicine, to the tumor ER, for the long-term adjuvant therapy of breast cancer patients was a life-saving strategy. This innovation with tamoxifen is now credited for extending the lives of millions of women worldwide. Subsequent advances in the development of AIs for the long-term adjuvant therapy of breast cancer in postmenopausal patients improved response rates and decreased the numbers of side effects noted with tamoxifen (thromboembolic events and endometrial cancer) and also prompted the use of a safer SERM, raloxifene, and the use of exemestane (an AI) for the chemoprevention of breast cancer in high-risk postmenopausal women.
| Therapy | ER+ | ER– | ER+/– |
|---|---|---|---|
| Castration | 25/33 | 4/53 | 0/2 |
| Adrenalectomy | 32/66 | 4/33 | 2/8 |
| Hypophysectomy | 2/8 | 0/8 | — |
| T otal | 55/107 = 55% | 8/94 = 8% | 2/10 = 30% |
| Estrogen | 37/57 | 5/58 | 0/2 |
| Androgen | 12/26 | 2/24 | 0/1 |
| Glucocorticoid | 2/2 | — | — |
| T otal | 51/85 = 60% | 7/82 = 8% | 0/3 = 0% |
a Out of the outcome of 459 treatment trials meeting the criteria of objective tumor response or failure, ER assays were available in 436 of these trials.
A Current View of the Molecular Mechanism of Estrogen Action
The ER is located in the estrogen target tissues around a woman’s body, including the majority of breast cancers. It is important to note that the ER is also present in high quantities in male breast cancer, so this mechanism will also apply in that context. The first ER discovered is now referred to as ERα. A second ER, known as ERβ, prompted an intense investigation of the role of both receptors in physiology and breast cancer. At present, ERα is considered to be the principal driver of ER-mediated breast cancer growth and the site of mutations responsible for acquired antihormonal resistance. An exact role for ERβ for breast cancer prognosis and therapy is yet to be defined.
The NSRs for consideration are illustrated in Fig. 21.3 , and their structural similarities are compared and contrasted. The domains are named from the N-terminus, A through F. The important area that allows communication of the ligand-activated ER with the promoter region of ER-responsive genes is described as the DNA-binding domain (DBD), or region C. The ligand-binding domain (LBD) is toward the C-terminus and is referred to as region E, which also encompasses the F region. Domain F modulates the function of the ER in a ligand-, promoter-, and cell-specific fashion. Deletion analysis of the ER has identified two regions with activating functions that are important for the biological activity of the estradiol (E2)-ER complex. Activation function (AF)-1 is in the A/B region of ERα, but this is a truncated region in ERβ (see Fig. 21.3 ). As a result, the E2-ERβ complex does not have the same estrogenic efficacy of the E2-ERα complex. Indeed, laboratory experiments demonstrate that ERβ can block the estrogen-like action of the E2-ERα complex. This is because, within the E2-ER complex, AF-1 and AF-2 collaborate with each other at the promoter region for full estrogen-like activity.
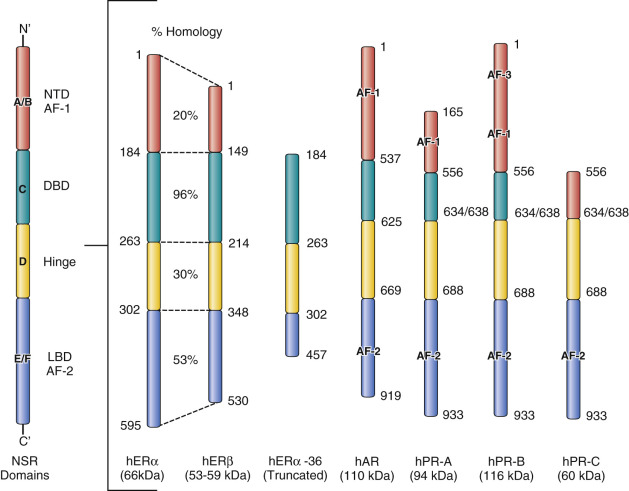
The LBD of ERα, has been studied in great detail and crystallized with numerous ligands, eg: estradiol, diethylstilbestrol, and the SERMs: 4-hydroxytamoxifen and raloxifene (see Fig. 21.2 ). The LBD of ERβ only differs to that of ERα by 2 amino acids. The amino acids Leu384 and Met421 in ERα are replaced by Met336 and Ile373, respectively, in ERβ. Despite the similarities in the LBD, considerable efforts continue to discover ERα and ERβ specific agonists and antagonists.
Estrogen action is triggered by the diffusion of E2 from the circulation into estrogen target tissues, where the ligand binds with high affinity K d = 10 –10 M within the unoccupied LBD of ERα. The 3′ phenolic hydroxyl of estradiol is tethered through amino acids glutamic acid (Glu353) and arginine (Arg354), and the 17′ alcoholic hydroxyl at amino acid histidine (His524). This interaction causes a conformational change in the ER with helix 12 sealing the ligand within the LBD. As a result, the external surface of the E2-ER complex exposes AF-2, which allows coactivator molecules (SRC-1, SRC-2, or SRC3) to bind and to construct a transcription complex at the promoter region of estrogen responsive genes. AF-1 and AF-2 cooperate in building the transcription complex. However, the transcription complex is not a static trigger but is required to “breathe” through the destruction of the complex by the proteasomal ubiquitination system (the process is illustrated for estradiol and fulvestrant action shown in Fig. 21.2 ). The promoter sites are recharged to continue the function of the transcription complex. In this way, the cell is guided through the cell cycle of checkpoints, protein synthesis, and division.
The central position of ER in breast cancer growth has identified this receptor protein as the preeminent target for the therapeutic agents referred to as SERMs and SERDs.
The Molecular Mechanism of Action of SERMs
The important pharmacologic property of the SERMs tamoxifen and raloxifene (see Fig. 21.2 ) is the different intrinsic estrogenic activity of the respective ER complexes. For example, tamoxifen-ER complexes are estrogenic and thus build bone in postmenopausal women and lower circulating low-density lipoprotein cholesterol (LDL). However, the promiscuous estrogenicity of the tamoxifen-ER complex that accumulates in the nucleus also increases the risk of estrogen-like side effects, such as thromboembolic events and endometrial cancer. Raloxifene has a short biological half-life because it is a polyhydroxylated compound in contrast to tamoxifen, which is lipophilic and heavily protein-bound. The raloxifene molecule is the active agent at the ER, but the tamoxifen molecule is required to be hydroxylated and demethylated to form 4-hydroxytamoxifen and endoxifen (see Fig. 21.2 ) before receptor binding. The argument has been made that mutations in the CYP2D6 gene impair tamoxifen’s efficacy; however, this is not proven, and clinical testing is not a standard of care.
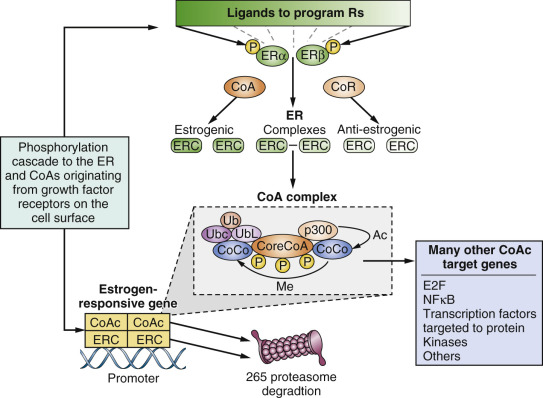
Early molecular pharmacology studies of the structure-function relationships of nonsteroidal antiestrogens related to tamoxifen demonstrated that the bulky antiestrogenic side chain of nonsteroidal antiestrogens prevented the sealing of the ligand within the LBD. This mechanism was confirmed by x-ray crystallography, which showed both 4-hydroxytamoxifen and raloxifene prevented helix 12 from closing. The SERM-ER complex can bind to the promoter region of estrogen responsive genes, but the conformation of the complex now prevents coactivator binding and the building of a transcription complex ( Fig. 21.4 ). The important aspect of SERM pharmacology is that the ligand binding within the LBD can alter the external surface of the ER complex, which in turn influences the proportion of coactivators or corepressors that can bind. These molecular maneuvers predetermine the balance of estrogen and antiestrogen-like properties of the SERM-ER complex at the target site (see Fig. 21.4 ).
Related posts:
 Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
 Therapeutic Strategies for Breast Cancer
Therapeutic Strategies for Breast Cancer
 Breast Conservation Therapy for Invasive Breast Cancer
Breast Conservation Therapy for Invasive Breast Cancer
 Radiation Complications and Their Management
Radiation Complications and Their Management
 Immunologic Approaches to Breast Cancer Therapy
Immunologic Approaches to Breast Cancer Therapy
 Delayed Diagnosis of Symptomatic Breast Cancer
Delayed Diagnosis of Symptomatic Breast Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


