Since 1975, the overall mortality rates for childhood cancer have decreased by 50% and survival rates for childhood cancer now exceed 80%; however, this progress has been most evident for children with hematologic malignancies, and mortality rates for children affected by some of the most common pediatric solid tumors such as neuroblastoma and sarcomas have remained virtually unchanged over the past 20 years.2 The reasons for the lack of progress in patients with solid tumors when compared to those with hematologic malignancies is unclear but might be related to the relatively limited numbers of patients available to conduct randomized trials and the scarcity of recurrent and actionable genetic aberrations reported to date.2,3
MULTIDISCIPLINARY CARE IS ESSENTIAL FOR CHILDREN AND ADOLESCENTS WITH SOLID TUMORS
The management of pediatric solid tumors is complex and requires a coordinated multidisciplinary approach that incorporates multiple subspecialties including surgery, radiology, pathology, radiotherapy, oncology, and physical therapy.4
Local control of the tumor may be achieved with the use of surgery, radiation therapy, or both. The use of neoadjuvant chemotherapy offers the theoretical advantage of decreasing the risk of micrometastatic dissemination as well as primary tumor size and, therefore, limiting the extent of surgery and the radiation doses.4 In addition, the administration of preoperative chemotherapy facilitates the evaluation of histologic response in the resected surgical specimen providing important prognostic and therapeutic information.5
Surgery plays a dual role in the management of pediatric solid tumors because it provides tissue for diagnosis and biologic studies as well as a mechanism for removing the primary tumor. The role of debulking surgery is controversial.6,7 Surgery can also be used later in the course of treatment to remove residual tumor, and the results of the surgery may provide important information regarding the need for additional radiotherapy or chemotherapy.8–10
The radiation oncologist should evaluate the patient prior to the initiation of any therapy and to help select the radiation plan and optimal treatment modality. The patient’s age, extent of disease, and disease status all play a role in selecting the appropriate radiotherapeutic approach from the available techniques including intensity modulated radiation therapy (IMRT), three-dimensional (3D) conformal radiotherapy, proton beam radiation therapy (PBRT), and brachytherapy.
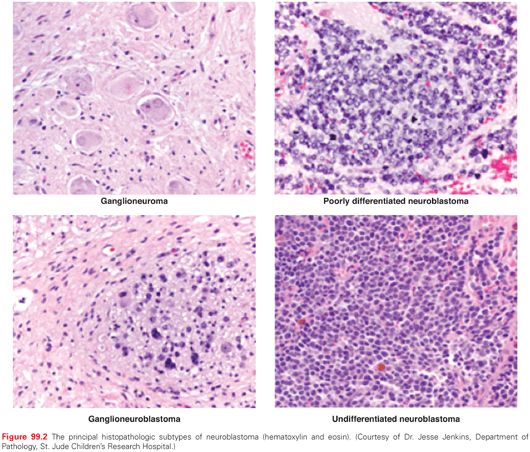
Neuroblastomic tumors comprise a spectrum of tumors that arise from primitive sympathetic ganglion cells and include neuroblastomas, ganglioneuroblastomas, and ganglioneuromas (Fig. 99.2). They can originate anywhere along the sympathetic ganglia; about half arise in the adrenal medulla. This group of tumors is known for a broad spectrum of clinical behaviors ranging from a completely benign mass to spontaneous regression to widely disseminated, aggressive, and often fatal disease.11–14 Tumors can be subdivided into distinct risk categories based on clinical and biologic features.11,15 Historically, treatment has been tailored based on determination of the risk of recurrence for a given patient. A combination of clinical and biologic features of disease have been used to classify patients as low, intermediate, and high risk.11,15
Epidemiology
Neuroblastoma is the third most common childhood cancer after leukemia and brain tumors. From 2006 to 2010, the age-adjusted incidence of neuroblastoma was 10.1 per 1 million children aged 0 to 14 years.1,16 During the first year of life, neuroblastoma accounts for the majority of cancer cases with an incidence almost double that of leukemia; 16% of infant neuroblastomas are diagnosed during the first month of life.16
There are approximately 650 children diagnosed each year with neuroblastoma in the United States16 and their median age at diagnosis is 19.1 months (573 days).17 Neuroblastoma is slightly more common in Caucasians and males, with the greatest sex-specific incidence difference occurring during infancy.16
The majority of neuroblastomas are sporadic. About 1% to 2% of cases are associated with a history of neuroblastoma in immediate or extended family members.18–23 An early analysis of these family pedigrees suggests an autosomal-dominant mode of inheritance with incomplete penetrance.24 Although many of these familial neuroblastoma patients present before 1 year of age, and more frequently have multifocal primaries,14,25 there is also great clinical and biologic heterogeneity among patients in the same kindred.19–21,26 Recently, germ-line mutations in the anaplastic lymphoma kinase (ALK) gene were identified as the cause of most cases of hereditary neuroblastomas.25,27–30 Another less common cause of hereditary neuroblastoma involves germ-line mutations of the pairedlike homeobox 2B (PHOX2B) gene.31–33 These children often have concomitant neural crest disorders (neurocristopathies),34 including Hirschsprung disease,32,34,35 and congenital hypoventilation syndrome.33,34,36 Other genetic conditions that may predispose for the development of neuroblastoma include neurofibromatosis,32,37,38 Turner syndrome,39 and Beckwith-Wiedemann syndrome.40
Pathology
Neuroblastic tumors, which include ganglioneuromas, ganglioneuroblastomas, and neuroblastomas (see Fig. 99.2), are derived from primitive cells involved in the development of the sympathetic nervous system.41 The most undifferentiated neuroblastic tumors are composed of neuroblasts with very few schwannian (stromal) cells. Immunohistochemistry may be useful in distinguishing neuroblastoma from other small round blue cell tumors, and immunoreactivity is commonly observed to neuron-specific enolase, synaptophysin, chromogranin A, NB84, and S100. As tumors become more differentiated, the ratio of Schwann to neuroblastoma cells increases, and the neuroblasts appear more mature. Ganglioneuroblastoma can be subdivided into either a stroma-rich, intermixed variant, or nodular variant. Ganglioneuroma is predominantly composed of Schwann cells studded with maturing or fully mature ganglion cells and is considered a benign tumor. These tumors were originally classified according to an “age-linked” classification system by Shimada et al.,42 which was subsequently clarified and adopted for international use by the International Neuroblastoma Pathology Committee (INPC).43–45 These tumors are morphologically classified into four histologic subtypes according to the balance between neural-type cells (primitive neuroblasts, maturing neuroblasts, and ganglion cells) and the Schwann-type cells (Schwannian blasts and mature Schwann cells) and the degree of surrounding Schwannian stroma: (1) neuroblastoma (Schwannian stroma-poor), undifferentiated, poorly differentiated, and differentiating; (2) ganglioneuroblastoma, intermixed (Schwannian stroma-rich); (3) ganglioneuroma (Schwannian stroma dominant), maturing and mature; and (4) ganglioneuroblastoma, nodular (composite Schwannian stroma rich/stoma dominant and stroma poor).43 The grade of neuroblastic differentiation and mitosis-karyorrhexis index (MKI; defined as the number of tumor cells in mitosis and in the process of karyorrhexis) and age of patient are then used to assign to either a favorable or unfavorable prognostic group (Fig. 99.3).44
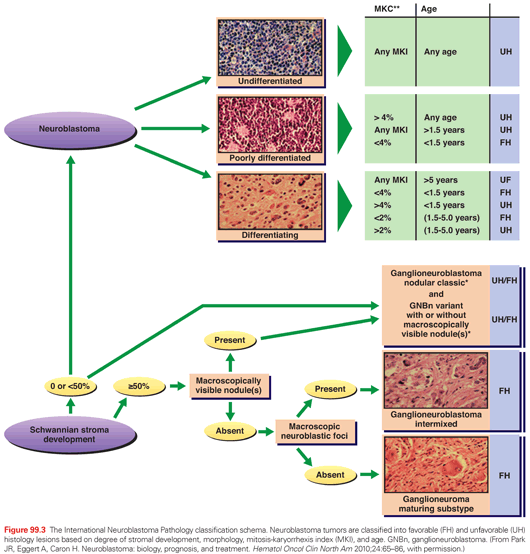
Neuroblastoma tumors are classified into favorable histology (FH) and unfavorable histology (UH) lesions based on the degree of stromal development, morphology, MKI, and age.
The INPC system’s prognostic significance was confirmed in a retrospective review of two Children’s Cancer Group (CCG) protocols that demonstrated a threefold difference in the 5-year event-free survival (EFS) for patients with FH versus UH features.46
Molecular Biology
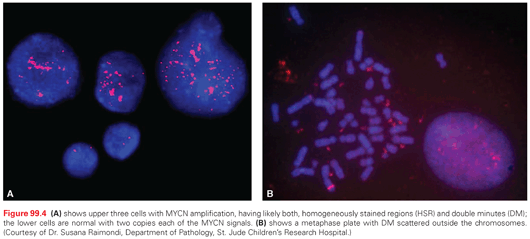
Amplified segments of DNA in the form of homogeneously staining regions of chromosomes (HSR) or extrachromosomal DNA called double-minute chromosomes (DMs) are often found in neuroblastomas (Fig. 99.4). The gene that was consistently amplified in these segments was identified by Schwab and colleagues47 as a myc-related gene, which was eventually called MYCN (v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog). Amplification of MYCN is predominantly associated with advanced stage disease, rapid tumor progression, and poor prognosis.48,49 A 5- to 500-fold amplification of the MYCN gene is found in about 22% of all patients with neuroblastoma,11 and in approximately 30% to 40% of patients with advanced stage disease (see Fig. 99.4).48,49 Amplification (increased gene copy number) results in persistently high levels of the MYCN protein, a DNA-binding transcription factor known to cause malignant transformation in tumor models. The negative prognostic significance of MYCN amplification (>10 copies per haploid genome) is independent of age, stage, or other genetic alterations and remains an important part of the current international risk group classification system.50,51
The DNA content (ploidy) of tumors is either near diploid or hyperdiploid.50,52 Near-diploid tumors in patients less than 18 to 24 months of age are associated with an adverse prognosis, whereas hyperdiploid tumors in this age group are associated with low-stage disease and a favorable outcome.53,54 In the new International Risk Group pretreatment classification system, ploidy is only used to stratify patients less than 18 months old with metastatic disease who do not have MYCN-amplified tumors.51
Chromosomal deletions or gains are also frequently identified. Loss of heterozygosity (LOH; loss of one allele) at chromosome 1p36 and 11q occurs in 23% and 34% of tumors respectively.55 A gain of chromosome 17 or 17q is present in 50% to 60% of neuroblastomas and is associated with an aggressive phenotype.56 Trk, a family of neurotropin receptors, is important in the development of the central and peripheral nervous system. Elevated expression of TrkA or TrkC is seen in favorable, low-stage tumors. Low expression of TrkA or elevated expression of full-length TrkB is seen in advanced-stage MYCN-amplified tumors.57 Additionally, TrkB expression has been linked to chemotherapeutic resistance.57,58 Among the most common mutations in sporadic neuroblastoma is in the α-thalassemia/mental retardation syndrome X-linked (ATRX) gene, which is seen in children over 12 years of age (44%).59 The evaluation of a chromosomal translocation in a neuroblastoma patient led to the identification of a family of genes on chromosome 1, now called neuroblastoma breakpoint family (NBPF).60 Copy number variation in this region (1q21.1) has also been shown to be highly associated with neuroblastoma, probably through disruption of one of the NBPF genes, NBPF23.61 A number of technologic advances have enabled the analysis of whole genome or exomes of tumors to provide insight into the pathogenesis of neuroblastoma and are under investigation to refine risk assignment.62–68 For example, risk polymorphisms at 6p22, identified by genome-wide association studies (GWAS), have been associated with sporadic neuroblastoma, and homozygosity for any of these alleles is associated with high-risk features.69 Similarly, common variations in the BRCA1-associated RING domain 1 (BARD1) gene have been implicated in the development of high-risk disease.70 By expanding these GWAS studies, this same group also identified risk alleles within the LMO1 (LIM domain only 1) gene at 11p15.4 that were more common in tumors with high-risk disease (p <0.0001), suggesting that LMO1 may be a neuroblastoma oncogene.71
Diagnosis
About two-thirds of neuroblastomas originate in the abdomen, with 50% beginning in the adrenal gland. Other common sites include the chest (16%), neck (3%), and pelvic sympathetic ganglia (3%). Approximately 50% of tumors have spread to the bone or bone marrow at presentation.51 The pattern of metastatic spread varies with age and histology of the tumor. Infants with favorable histologies are more likely to have liver and skin metastases. Unfavorable histology tumors are more likely to spread to the bone marrow and bones.72 At presentation, abdominal tumors can cause increased abdominal girth, a palpable mass, diarrhea, or constipation. Many thoracic tumors are detected incidentally; however, large thoracic or cervical tumors can cause Horner syndrome, superior vena cava syndrome, or mechanical airway obstruction. Children with bone metastases may have a limp, complain of pain, or develop periorbital swelling, ecchymoses, or proptosis. Fever and hypertension are often present.
The evaluation of a child suspected of having neuroblastoma begins with a careful history and physical examination. A detailed head and neck exam, looking for skull metastases; “raccoon eyes,”73 proptosis, or other eye abnormalities; and Horner syndrome should be performed. Attention to neurologic function is critical due to the paraspinal location of many tumors, which can result in spinal cord compression and permanent loss of function if not treated expeditiously.74 Similarly, blindness has developed in children with periorbital disease while staging evaluations were being completed.75 In infants, the examination must include a search for skin and subcutaneous metastases that appear as reddish-purple, raised lesions.76 Because neuroblastomas produce catecholamine metabolites, the measurement of these can often help narrow the diagnosis.77,78 Urine samples for quantitative excretion of vanillylmandelic acid (VMA) and homovanillic acid (HVA) are elevated in more than 80% of patients79 and should be part of the diagnostic evaluation for every patient. These markers are also useful to evaluate the effectiveness of therapy and to monitor for disease recurrence.80 Ultrasound is often the initial radiologic study to identify the tumor. However, magnetic resonance imaging (MRI) or computerized tomography (CT) of the chest, abdomen, and pelvis is required to clearly define the location and extent of the primary tumor. Radiographically, a neuroblastoma typically appears as a heterogeneous mass with calcifications. Adrenal and retroperitoneal tumors characteristically involve and displace the major vessels.
Patients with paraspinal primary tumors may have asymptomatic extension through the spinal foramina. Due to the increased risk of neurologic compromise, these patients should undergo further imaging of the spinal canal using MRI with contrast. Imaging of the head should be considered in any child with palpable skull lesions, ptosis, or orbital ecchymosis.75
Tumors can secrete a vasoactive intestinal polypeptide resulting in intractable diarrhea and abdominal distention that resolves after resection. In the largest review of 22 patients, most presented with significant weight loss and metabolic abnormalities, and 16 out of 22 had 1 to 21 months of gastrointestinal (GI) symptoms prior to the diagnosis of neuroblastoma. Although usually associated with differentiating tumors of low stage, 6 of these 22 were high-risk patients who developed diarrhea upon initiation of chemotherapy.81 Opsoclonus-myoclonus-ataxia (OMA) syndrome, often called dancing eyes,82 dancing feet is seen in up to 3% of children with neuroblastoma and presents with chaotic, multidirectional eye movements, spontaneous limb jerking, and ataxia. Many patients also have significant behavioral problems, sleep disturbances, and learning defects.83,84 Although this process is thought to arise as a consequence of immune-mediated cross-reactivity between neuroblasts and the central nervous system, no specific antibody or lymphocyte marker has been identified.85 The tumors associated with OMA patients typically are of favorable biology and limited stage, and thus, patients with this condition have an excellent cancer prognosis.86,87 Although the movements may resolve over time, most children have persistent neurologic deficits, including speech and cognitive delays and behavioral problems.88,89
Spinal cord involvement with infiltration of the intervertebral foramina is often seen in the initial evaluation of neuroblastoma90; however, only about 5% of children at the time of diagnosis develop symptoms related to spinal cord compression.74,91 Spinal cord compression is considered an oncologic emergency because prolonged compression of the spinal cord can lead to irreversible loss of function. This can be treated with urgent radiation therapy, neurosurgery, or chemotherapy. Although there is no current consensus as to the best initial treatment,92 a retrospective analysis suggests that each of these modalities are equally effective in the short term. However, there are significant differences in late effects.90,93–95 The management of this rare presentation requires an experienced multidisciplinary team and the treatment recommendations should be made on a case-by-case basis specific to the severity and duration of symptoms and an assessment of the anticipated short- and long-term risks of the specific intervention.90,92
Metaiodobenzylguanidine (MIBG) is an analog of norepinephrine and is taken up by tissues that express the norepinephrine transport protein, which includes almost 90% of neuroblastomas.96,97 With a sensitivity of about 90% and a specificity of 100%, MIBG has replaced technetium-99m bone scans and has been recommended as a standard agent for staging patients with neuroblastoma96,98,99 because of its sensitivity and specificity; all patients should undergo MIBG scans at the time of diagnosis and at subsequent intervals after the start of treatment to assess response. In the 10% of patients who have negative MIBG scans, fludeoxyglucose (18F-FDG) positron-emission tomography (PET) scans can be considered.98 Newer PET agents, such as fluorine-18-L-dihydroxyphenylalanine (18F-Dopa) are also under investigation.100 Bone marrow involvement should be further evaluated by bilateral aspiration and a biopsy.99,101
Other malignancies for which neuroblastoma can occasionally be confused include other small, round, blue cell tumors such as rhabdomyosarcoma, the Ewing sarcoma family of tumors, non-Hodgkin lymphoma, and acute leukemia. If the diagnosis is to be based on material from bone marrow, it is recommended that these cells be confirmed to be neuroblasts by immunohistochemistry staining with synaptophysin102 or chromogranin103 and that urine HVA and/or VMA be >3.0 SD above mean per milligram creatinine for age.99
Screening
Because infants have the best prognosis and because more than 80% of patients with neuroblastoma have elevated catecholamine markers that can be easily measured, screening infants was thought to be a promising approach.104 Unfortunately, two carefully controlled trials definitively demonstrated that screening did not reduce mortality and, in fact, because many infant neuroblastomas spontaneously regress, led to over diagnosis and treatment of patients that needed no intervention.105–108
Staging
The International Neuroblastoma Staging System (INSS), developed in 1988109 and revised in 199399 (Table 99.1) was developed to facilitate comparison among trials and is the current staging system used in North American and European cooperative group trials. This system is a postsurgical staging system, in that it uses the extent of initial surgical resection to stage patients. The INSS is significantly affected by the experience of the surgeon, leading to variability in stage assignments. To better define homogenous pretreatment patient cohorts and compare trials conducted in different regions of the world, another staging system, based on preoperative, image-defined risk factors (Tables 99.2A and Tables 99.2B), called the International Risk Group classification system.110 The predictive prognostic significance of this system will be tested in future prospective clinical trials.


Management by Risk Group
The treatment of neuroblastoma incorporates a number of clinical and biologic variables, in addition to the stage of disease, to evaluate the risk of recurrence and to thus determine the intensity of treatment. The Children’s Oncology Group’s (COG) risk categories incorporate the patient’s age at presentation, stage of the tumor, histologic appearance, quantitative DNA content, and presence or absence of MYCN amplification within the tumor cells (Table 99.3).
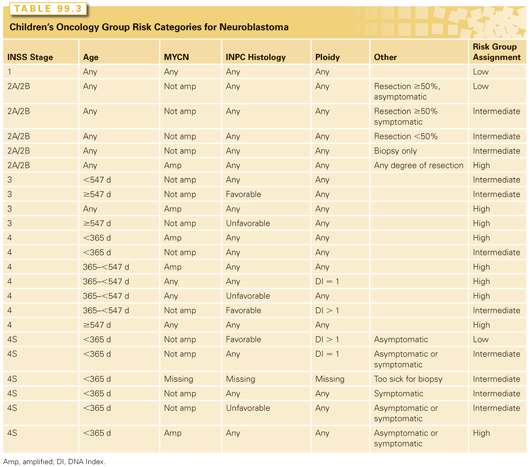
The age at presentation is an important prognostic factor, because infants tend to have tumors with more favorable features, including stage and histology. A multivariate analysis of 3,666 patients enrolled in cooperative group studies demonstrated 82% 4-year EFS for children less than 18 months versus 42% for those more than 18 months.17 Adolescents and young adults rarely present with neuroblastoma, but their tumors tend to be indolent and fatal.110–112
The variables selected by other pediatric oncology cooperative groups to define the risk categories have not been uniform. To be able to compare trials worldwide, an international conference developed a consensus approach to pretreatment risk stratification, now called the International Neuroblastoma Risk Group (INRG) Classification System (see Tables 99.2A and Tables 99.2B).51 This new classification system is different than that of the COG in that it uses the new INRG staging system,113 and adds the presence/absence of 11q aberrations. As new trials are developed, the value of this new risk group classification system will be validated.
Low Risk
Children with low-risk neuroblastomas generally include patients of any age with localized, resectable tumors (Table 99.4). Low-risk tumors comprise about 50% of all neuroblastomas. Surgery is the primary treatment, and overall survival rates are greater than 90%.114–116 Recently, a prospective study of 87 infants less than 6 months of age and with small (volume ≤16 mL) isolated adrenal masses who were observed only showed that 81% (67 out of 83) were spared surgery with a median follow-up of 3.2 years and an overall survival at 3 years of 100%.117 Thus, expectant observation without surgery in this subgroup of patients should be considered standard treatment and, in fact, the authors recommend extending this approach to “all localized noninfiltrative neuroblastoma tumors [corresponding to the International Neuroblastoma Risk Group Staging System stage L1; see Table 99.2]113 in infants less than 1 year of age.”117
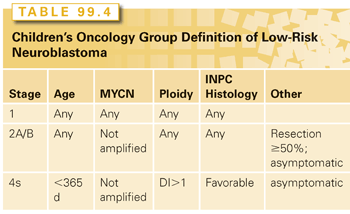
A 4S neuroblastoma is a special118 entity because the high rate of spontaneous regression allows for a delay or elimination of chemotherapy for up to 80% of children.119,120 Two subsets are more likely to require chemotherapy: those tumors with unfavorable biologic features (i.e., MYCN amplification) and tumors in infants younger than 2 months who have extensive liver disease. Untreated, the latter case leads to a higher incidence of mortality, because massive hepatomegaly prevents adequate chest wall expansion.120,121 These children should receive chemotherapy or low-dose radiation (4.5 to 6 Gy in three to four fractions) to decrease tumor size.
Intermediate Risk
Children with intermediate-risk tumors comprise 10% to 15% of new cases and are defined as: infants (<365 days old) with INSS 3 or 4 tumors without MYCN amplification and favorable histology, children (≥365 days old) with INSS 3 tumors without MYCN amplification and favorable histology, or INSS 4 with unfavorable histology and/or diploid DNA (Table 99.5).122 Surgery is an important component of treatment, as is the addition of moderately intensive chemotherapy (i.e., carboplatin, etoposide, adriamycin, cyclophosphamide). Radiotherapy is reserved for children with progression or those who have unresectable tumors after chemotherapy. This approach has led to excellent survival of 50% to 100%, depending on the clinical and biologic characteristics.123–125 In a recent report from the COG, 479 children were treated according to a biologically based risk assignment system. Of these, 323 with favorable biologic characteristics received four courses of chemotherapy and 141 children with unfavorable biology received eight courses. This approach resulted in a 3-year EFS of 88±2% and 3-years overall survival of 96±1%.122
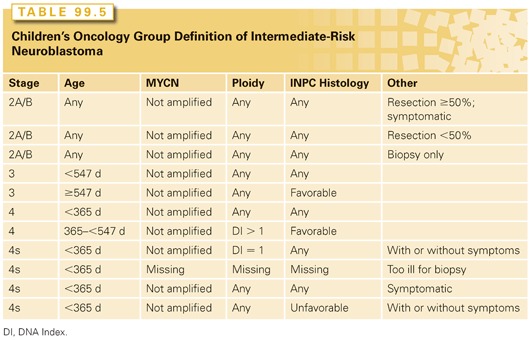
High Risk
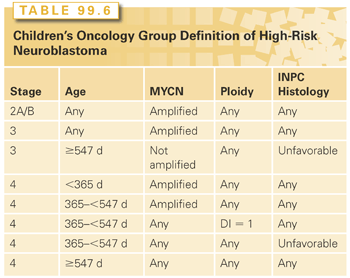
Approximately 50% to 60% of children present with metastatic disease at diagnosis and the vast majority of these are classified as high-risk patients.126 As seen in Table 99.6, children with high-risk disease predominantly include children more than 18 months of age with INSS 4 disease. Other subsets of high-risk patients include children of any age with INSS 2a/b or 3 disease and MYCN amplification, children ≥18 months with INSS 3 and unfavorable histology, and infants (<1 year of age) with MYCN amplification. The treatment consists of four general components: (1) induction chemotherapy, (2) surgical resection of all gross disease, (3) consolidation, which generally includes myeloablative chemotherapy with stem-cell rescue and radiation therapy to the tumor bed, and then (4) treatment of minimal residual disease (MRD). Despite this intensive therapy, almost half of all high-risk patients eventually relapse and die of their disease.126
Induction Chemotherapy. Standard chemotherapy regimens for high-risk disease include combinations of the epipodophyllotoxins etoposide or teniposide, platinum-based agents, cyclophosphamide, and doxorubicin; other active agents such as ifosfamide and topotecan have also been included in some regimens.127–134 A retrospective analysis in 1991 of 44 published clinical trials showed a convincing correlation between dose intensity and response and progression-free survival, particularly with cisplatin and teniposide.135 Although a later review did not corroborate these findings,136 the general approach worldwide has been to increase the intensity of induction chemotherapy. In the only recent randomized trial comparing induction regimens, 262 patients were assigned to either rapid treatment (N = 130) with cisplatin (C), vincristine (O), carboplatin (J), etoposide (E), and cyclophosphamide (C) (COJEC), or standard treatment (N = 132) with vincristine, cisplatin (P), etoposide, cyclophosphamide (OPEC) alternating with vincristine, carboplatin, etoposide, cyclophosphamide (OJEC). The rapid regimen was given every 10 days and the standard regimen was given every 21 days. The same cumulative doses of each drug, except vincristine, were administered and the relative dose-intensity of the rapid regimen was 1.94 compared to the standard regimen.137 There was no difference in the overall survival (OS) at 5 and 10 years, although the EFS at 5 years was significantly better in the rapid group (30.2% versus 18.2%; p = 0.022). Also, patients progressed to consolidation a median of 55 days earlier, prompting the authors to speculate that this might have contributed to the better outcome in the rapid group.137 The lack of difference in OS and relatively inferior outcomes, compared to other induction regimens,133 make interpretation of these results difficult. What is clear is that response, as measured by MIBG scans at the end of induction, strongly correlate with overall outcome138–140 and that further refinement in induction therapy may focus on those ultra high-risk patients who have inferior early response as measured by MIBG or more sensitive quantitative methods.141,142
Surgery. The role and timing of surgery in the management of children with high-risk neuroblastoma is uncertain.143 Although some reports of patients with stage 3 or 4 disease have found that gross total resection of the primary tumor and metastatic local–regional disease has been associated with improved local tumor control and increased overall patient survival,144–147 other reports have not.7,148,149 Nevertheless, the COG as well the major European cooperative group, SIOPEN,137 currently recommend gross total resection of the primary tumor and regional disease in patients with high-risk neuroblastoma. The resection is often delayed until near the end of induction, even though tumor volume reduction appears to plateau after the second or third course of chemotherapy,150 and there are some data that early resection may provide a survival benefit.151 The goal of surgical local control is to remove all visible tumors without jeopardizing vital organs or delaying chemotherapy; however, most primary tumors invade local structures and surround major blood vessels, making resection difficult. Complete removal at the time of diagnosis is possible for less than 20% of patients.146,152 Resection of invasive tumor is associated with complications, including the removal of normal organs, hemorrhage, renal injury,153 vascular injury, chylous ascites and chronic diarrhea. After treatment with chemotherapy, complete removal becomes possible in more than 50%.7
Consolidation. Early studies using high-dose chemotherapy showed a strong correlation of increasing dose intensity with progression-free survival in metastatic neuroblastoma.135,154 The critical importance of dose intensity ultimately led to the integration of high-dose chemotherapy and autologous stem cell transplant (ASCT) into modern treatment regimens for high-risk neuroblastoma.155–157 Although with it comes unique challenges and risks, the use of myeloablative ASCT allows for the delivery of chemotherapeutic doses not usually feasible due to the dose-limiting toxicity of myelosuppression. Although there is no consensus on the optimal conditioning regimen, many nonrandomized studies supported the use of ASCT as consolidation158 and several randomized trials have subsequently made ASCT following intensive induction chemotherapy part of the standard of care in the treatment of children with high-risk neuroblastoma.129,158–160
One of the most common myeloablative regimens used worldwide is a combination of busulfan and melphalan (BuMel). A recent retrospective analysis of nearly 3 decades of European experience with ASCT for neuroblastoma revealed a survival benefit for BuMel over other melphalan-based conditioning regimens.161 The 343 patients treated with this regimen in first remission had a 5-year OS rate of 48%.162 Specifics regarding transplant-related complications were not reported. A recent, smaller retrospective study from Spain using a BuMel conditioning reported a progression-free survival of 57% among 36 patients at a mean follow-up of 55 months.162 There were no toxic deaths reported and no reported veno-occlusive disease (VOD). Risk of complications from this regimen may be age dependent; a report of infants treated with a busulfan-melphalan regimen (with or without cyclophosphamide) reported VOD in 9 of 12 children treated, including one death.163 However, this regimen utilized oral busulfan, which has been shown to have a higher risk of complications.
Another common myeloablative regimen is a combination of carboplatin or cisplatin, etoposide, and melphalan (CEM), and it has been used in some of the largest reported US trials.160 The SIOP Europe Neuroblastoma Group (SIOPEN) randomized patients to receive BuMel or CEM in the HR-NBL1 trial with the primary aim to demonstrate superiority based on EFS. This randomization was closed early due to superiority of the BuMel regimen. A significant difference in EFS in favor of BuMel (3-years EFS 49% versus 33%) was observed as well as for OS (3-years OS 60% versus 48%; p = 0.004). This difference was mainly related to the relapse and progression incidence, which was significantly (p =0.001) lower with BuMel (48% versus 60%).164 Although these data clearly show the superiority of the BuMel conditioning regimen over CEM when used with the Rapid COJEC induction regimen of HR-NBL1,137 it remains to be demonstrated if this would hold true if BuMel were used after other induction regimens.
Because undetected neuroblastoma cells present in autologous bone marrow can contribute to relapse,165 the COG evaluated the role of purging stem cell products with a cocktail of five monoclonal antibodies targeting neuroblasts, attached to magnetic beads. All children received identical induction chemotherapy and myeloablation. Enrollees were randomly assigned to receive nonpurged peripheral blood stem cells (PBSC; N = 243) or purged PBSC (N = 243); 192 from the nonpurged and 180 from the purged group received ASCT. The 5-year EFS was 36% in the unpurged group (95% confidence interval [CI] 30 to 42) and 40% (33 to 46) in the group who received purged PBSCs (p = 0.77).166 Thus nonpurged PBSCs are acceptable for support of children receiving myeloablation during consolidation.
Based on improvements in EFS seen in patients treated with myeloablative consolidation and ASCT, several nonrandomized studies, which were further dose intensified using tandem, nonoverlapping myeloablative conditioning regimens. suggested this approach may further improve outcome.167–172 A pilot study of tandem transplant following intensive induction chemotherapy was developed by COG (ANBL00P1) to test the feasibility and toxicity for use as one of the arms of a randomized phase III study.172 Patients received the first consolidation with thioTEPA and cyclophosphamide and, upon recovery, received a second ABMT with carboplatin, etoposide phosphate, and melphalan. Forty-one patients were enrolled; eight did not receive any ASCT. Of the 33 who received the first ASCT, 26 went on to receive the second. The 3-year EFS was 44.8±9.5% and this tandem ASCT combination was used as the experimental arm in a randomized phase III trial comparing single versus tandem ASCT (ANBL0532).172 This trial completed accrual in February of 2012 and results are still pending.
Radiation Therapy. ASCT for neuroblastoma has been performed using various combinations of chemotherapy with and without total body irradiation (TBI). Although the optimum chemotherapeutic combination has not been conclusively demonstrated, the incorporation of TBI has not improved survival, but rather increases transplant-related complications.161,173 However, because primary site failure persists as a component of tumor progression, primary site irradiation has been used as part of consolidation therapy for patients with high-risk neuroblastoma,129,174–177 including those enrolled on the recent COG ANBL0532 study.178 In the setting of recurrent or incurable disease, radiotherapy can also be used to treat bone and soft tissue metastasis to control pain and to prevent a loss of organ function.179
Treatment of Minimal Residual Disease. Finally, additional treatment with biologics (i.e., 13-cis-retinoic acid) in the setting of minimal residual disease appears to improve survival. CCG randomized children after consolidation with high-dose chemotherapy and ASCT to receive 6 months of cis-retinoic acid. The 3-year EFS was significantly better among those who received cis-retinoic acid than those assigned to receive no further therapy (46% versus 29%, respectively; p = 0.027).129 Targeted immunotherapy, using a chimeric monoclonal antibody, ch14.18, against the tumor-associated disialoganglioside, GD2, has been ongoing for more than 2 decades. A recent randomized study from the COG found that the administration of ch14.18, in combination with interleukin-2, granulocyte macrophage colony stimulating factor, and cis-retinoic acid following recovery from consolidation was associated with an improved 2-year EFS (66±5% versus 46±5%) and OS (86±4% versus 75±5%) (p = 0.02).180 This study has established a new standard of care for all children with high-risk disease in the future.
Wilms tumor is the most common primary malignant renal tumor of childhood. Advances in therapy since the inception of the prospective randomized trials conducted by various multi-institutional cooperative groups have led to an OS rate of 90%, although success has been achieved at a cost of serious chronic health conditions 25 years after diagnosis in 25% of survivors.181
Epidemiology and Genetics
Among North American children less than 15 years of age, the incidence of Wilms tumor is 1 in 10,000 children, or approximately 500 new cases per year.182 Wilms tumor accounts for 6% of all childhood tumors, but more than 90% of all renal cancers in patients under the age of 20 years.183 The risk for developing Wilms tumor is higher in African Americans and lower among Asian populations.183 Although unilateral disease is more common, with males presenting at a slightly earlier age (37 months) than females (43 months), approximately 6% of patients harbor bilateral disease at diagnosis, with males presenting slightly earlier (24 months) than females (31 months).184
A variety of syndromes and congenital abnormalities are associated with the development of Wilms tumor. A deletion of chromosome 11p13 that encompasses the WT1 and PAX6 gene results in Wilms tumor, aniridia, genitourinary anomalies, and mental retardation (WAGR syndrome), whereas WT1 point mutations at 11p13 are implicated in Denys-Drash syndrome (DDS), and Frasier syndrome.185–187 A second Wilms tumor locus, WT2, is located at 11p15 and has been linked to an increased incidence of Wilms tumor in Beckwith-Wiedemann syndrome (BWS).186,188 Specific to the WT2 locus, at least 10 imprinted genes that have preferential expression by the maternal or paternal allele have been identified, including IGF2, H19, p57, and LIT1.186 Other overgrowth syndromes associated with an increased susceptibility for the development of Wilms tumor include Simpson-Golabi-Behmel syndrome, Sotos syndrome, and Perlman syndrome. Patients with isolated hemihypertrophy, aniridia, Bloom syndrome, Alagille syndrome, trisomy 18, or Li-Fraumeni syndrome are also at risk for developing Wilms tumor.186,189,190 Familial predisposition accounts for 1% to 2% of cases, and putative familial Wilms tumor genes have been mapped to 17q12-21 (FWT1) and 19q13.4 (FWT2).185 Children with hereditary syndromes that predispose to the development of Wilms tumor should receive abdominal screening ultrasounds every 3 months until 8 years of age.190 Although they appear to respond to treatment similarly as other children, patients with a predisposition (WT1 mutation) and Wilms tumor have a higher incidence of bilateral disease, intralobar nephrogenic rests, and end-stage renal disease.187
Pathology
Most Wilms tumors are solitary and composed of varying proportions of blastemal, stromal, and epithelial cells that recapitulate normal kidney development. Anaplasia, either focal or diffuse, is characterized by the presence of large nuclei, irregular mitotic features, and hyperplasia and is associated with adverse clinical outcomes.191,192 In focal anaplasia, cells with anaplastic nuclear changes are confined to sharply restricted foci within the primary tumor.192 A recent analysis has demonstrated that the presence of anaplasia even in stage I disease adversely affects clinical outcome.193
Wilms tumor must be distinguished histologically from other pediatric renal tumors, including renal cell carcinoma,194 clear cell sarcoma,195 and rhabdoid tumor of the kidney,196 as well as neuroblastoma. Clear cell sarcoma and malignant rhabdoid tumor of the kidney were previously considered to belong to the unfavorable histology group of Wilms tumors, but are now considered to be distinct categories of renal tumors. Rhabdoid tumor of the kidney tends to metastasize to the lung and brain. Primary rhabdoid tumors of the kidney and brain (atypical teratoid or rhabdoid tumors) share deletions of chromosome band 22q11.2, the site of SMARCB1 (also known as INI1, BAF47, and SNF5), a putative tumor suppressor gene.197,198 Another important renal neoplasm, congenital mesoblastic nephroma, is important to recognize because it is usually curable by nephrectomy alone. These tumors are typically identified in the first months of life, with a median age at diagnosis of 2 months.199 They are associated with the t(12;15) translocation that produces the ETV6-NTRK3 fusion gene product.200
Precursor lesions, or nephrogenic rests, have been identified in the renal parenchyma of approximately 36% of patients with Wilms tumor.201,202 Nephroblastomatosis is a term that describes kidneys with multifocal or diffuse nephrogenic rests. Perilobar rests, located along the perimeter of a renal lobe, are strongly associated with synchronous bilateral Wilms tumors, BWS, and hemihyperplasia, whereas intralobar rests (within the renal lobe) show a strong association with metachronous tumors and WAGR and DDS.203 Children with nephroblastomatosis have an increased risk of developing Wilms tumor and require close monitoring.204
Biologic prognostic factors, such as gain of 1q, LOH at chromosomal regions 1p and 16q, supplement stage, and histology when assigning risk. Gain of 1q is associated with increased risk of relapse in patients with favorable histology Wilms tumor.205 LOH of chromosomes 1p and 16q is present in approximately 5% of favorable histology tumors and is associated with increased risk of relapse and mortality.206 The recently closed COG trial incorporated these factors prospectively into treatment decisions with augmented therapy for patients with LOH and favorable histology; a data analysis is pending. In a retrospective analysis of patients with very low risk disease (stage 1, FH, <550 g, <24 months of age) who were treated with surgery alone, WT1 mutations, and 11p15 LOH were associated with a higher risk of relapse.207
Clinical Presentation and Natural History
Most children with Wilms tumor come to medical attention because of abdominal swelling or the presence of an abdominal mass that may be noted by the caregiver during bathing or dressing the child. Abdominal pain, gross hematuria, and fever may be present at diagnosis.208 Hypertension is present in approximately 20% of cases.209
Clinical Evaluation and Staging
With gentle palpation to maintain tumor capsule integrity, location and size of the abdominal mass and its movement with respiration should be noted. A varicocele may be associated with the presence of a tumor thrombus in the renal vein or inferior vena cava.210 An evaluation for signs of associated syndromes, such as aniridia, hemihypertrophy, and genitourinary abnormalities, is mandatory. Laboratory studies should include a complete blood cell count, liver function tests, renal function tests, serum chemistries including calcium, and a urinalysis.
Diagnostic Imaging
Imaging should define the local and distant extent of the tumor, evaluate the renal veins and surrounding vessels to detect the presence of a tumor thrombus, and assess the contralateral kidney to rule out bilateral lesions, all of which are important prior to surgery.
The initial radiographic study is often an abdominal ultrasound examination to determine the consistency of the mass and the organ of origin, and to assess patency of the inferior vena cava. CT or MRI is appropriate for imaging of the primary tumor and to provide adequate visualization of the contralateral kidney, liver, and abdomen to define metachronous and metastatic tumors in the abdomen and pelvis. MRI may be superior to other imaging modalities in delineating nephroblastomatosis.211
Both plain radiographs of the chest (chest x-ray) and a chest CT should be performed to determine the presence of pulmonary metastases. In many cases, nodules detected by more sensitive CT scans do not necessarily represent a metastatic tumor, and current studies focused on determining which patients truly require intensified therapy with radiation.212 Imaging of the brain or skeleton is usually reserved for patients with rhabdoid tumors of the kidney and clear cell sarcoma of the kidney.
Staging
The staging criteria developed by the National Wilms Tumor Study Group (NWTSG) is shown in Table 99.7. The NWTSG approach allows for an adequate assessment of the extent of disease and histologic characteristics of the tumor and facilitates the collection of tumor tissue for biologic studies prior to therapy. In contrast, the SIOP approach (staging not shown) uses preoperative chemotherapy, decreasing the volume of tumor, and thereby decreasing the perioperative risk of tumor spillage. As a result of these treatment philosophies, children enrolled on NWTSG trials receive radiation therapy more often, whereas patients on the SIOP trials receive more cumulative doses of anthracyclines.213–215
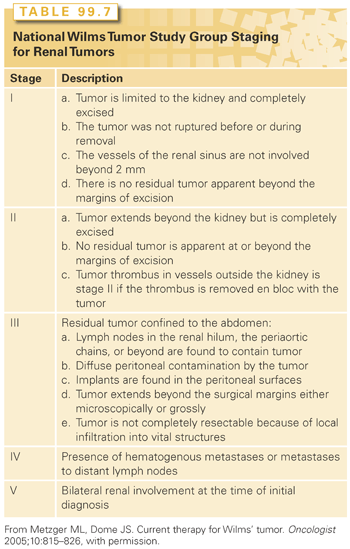
Treatment
Tumor stage and histology are the main prognostic indicators that determine the treatment regimen for Wilms tumor, whereas patient age at presentation and biologic factors (LOH 1p and 16q) were tested prospectively in the most recent COG (NWTS) trials (results pending). Surgery and chemotherapy comprise the main therapy for Wilms tumors, with the addition of radiation for metastatic or histologically aggressive tumors.
Surgery
Surgical resection is the primary method for achieving local control and is usually performed at the time of diagnosis in North American studies (COG/NWTS). A review of children treated on the NWTS-IV demonstrated an increased incidence of local recurrence in those cases in which lymph node biopsies were not obtained.216 Presumably, these children were understaged and thus undertreated. Additionally, this review clearly demonstrated that operative rupture, whether localized to the renal fossa or diffusely spread within the peritoneal cavity, was associated with an increased incidence of local recurrence.216
Routine exploration of the contralateral kidney is not necessary if imaging is satisfactory and does not suggest a bilateral process.217,218 Nephrectomy alone followed by observation for patients less than 2 years of age with stage I favorable histology tumors that weigh less than 550 g resulted in only an 85% EFS, and even though the study was stopped early, the OS was still 100% for this group of patients.219,220 Further analysis identified a WT1 mutation and an 11p15 LOH as risk factors associated with relapse in this patient group.207 Prenephrectomy chemotherapy and nephron sparing techniques may benefit patients with bilateral disease at diagnosis.221 Children with syndromic, unilateral Wilms tumor who have an increased risk of late renal failure (i.e., DDS or WAGR) may also benefit from partial nephrectomy or nephron sparing surgery.222,223 Preoperative treatment of Wilms tumor should be considered in children with a solitary kidney, bilateral renal tumors, tumor in a horseshoe kidney, tumor thrombus in the inferior vena cava above the level of the hepatic veins,224,225 and respiratory distress as a result of extensive metastatic disease.226 In children with bilateral disease or involvement of a solitary kidney, preoperative chemotherapy is intended to permit maximal conservation of uninvolved renal parenchyma. In the current COG renal tumor protocol, children who present with bilateral renal masses receive two to four cycles of chemotherapy without biopsy (COG-AREN0534, NCT00945009). A biopsy is reserved for those patients whose tumors do not show appropriate volume reduction. Definitive surgery should be performed by week 12.
The role of surgical resection in the management of solitary pulmonary metastases was evaluated by the NWTSG in 211 patients with no therapeutic benefit identified when surgical resection was added to pulmonary radiotherapy and chemotherapy.227 Four-year survival rates were identical in the two groups.
Chemotherapy
Vincristine and dactinomycin, with the addition of doxorubicin for more advanced disease, form the backbone of most combinations for the treatment of Wilms tumor.213 The recommended standard drug regimens and duration of therapy according to stage and intergroup studies are listed in Table 99.8. The five consecutive NWTSG studies have provided the foundation for current care in North America and the results of various US and European trials are depicted in Table 99.9.222,228–233,236,237
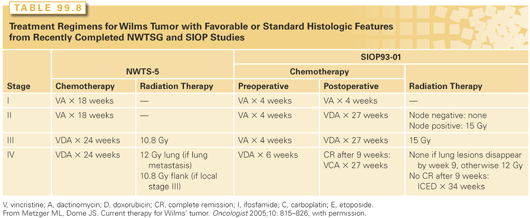
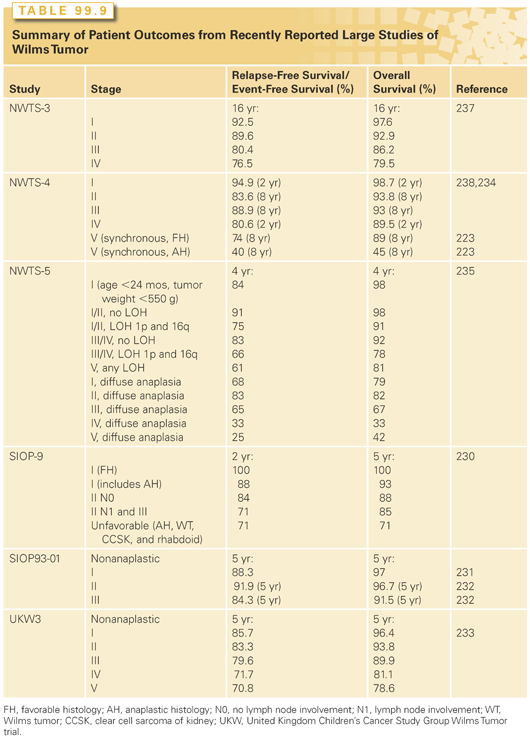
NWTS-1 documented that postoperative radiation therapy is not required for children with stage I/II favorable histology or stage I anaplastic histology tumors when postnephrectomy chemotherapy with vincristine and dactinomycin is administered; NWTS-2 showed that doxorubicin improved relapse-free survival for group II through IV patients, NWTS-3 upstaged all patients with lymph node involvement to stage III, and provided evidence that patients with stage III favorable histology who receive adjuvant chemotherapy and radiation require augmented therapy with either doxorubicin or higher radiation therapy dose (20 Gy) for successful outcomes. Concerns about late effects of radiation resulted in the selection of the three-drug regimen (vincristine, dactinomycin, and doxorubicin) with lower radiation therapy dose (10 Gy) as standard therapy in this group.234,235 Furthermore, this trial showed that the addition of cyclophosphamide to the standard three-drug regimen (vincristine, dactinomycin, and doxorubicin) does not improve the outcome of patients with stage IV/favorable histology tumors, but was potentially useful in patients with anaplastic tumors. In NWTS-4, the addition of cyclophosphamide proved to benefit patients with stage II through IV anaplastic tumors. In addition, therapy was reduced for low-risk patients, and pulse-intensive regimens were shown to maintain excellent survival with less toxicity than previous regimens.236 Anaplasia, either diffuse or focal, adversely affects the outcome even after the administration of conventional chemotherapy with vincristine, dactinomycin, and doxorubicin. Cyclophosphamide and etoposide, when added to vincristine, dactinomycin, and doxorubicin, improved the survival of patients with stage II through IV diffuse anaplasia in NWTS-5.193 A trial incorporating carboplatin to improve outcomes was stopped early due to toxicity of this regimen (UH1). Results of NWTS-5 indicate that patients with stage I anaplasia had lower than expected survival and will require augmented therapy in upcoming studies.
Salvage therapy is successful in up to 80% of patients with low-stage favorable histology tumors initially treated with vincristine, dactinomycin, and no radiotherapy.237 Late recurrence more than 5 years after the diagnosis is rare, but is associated with a similar outcome to earlier recurrence.238 At the time of relapse, approximately 50% of unilateral Wilms tumor patients who have received standard chemotherapy, three-drug chemotherapy, and radiation can be successfully re-treated.239–241 Alternating cyclophosphamide/etoposide and carboplatin/etoposide, surgery and radiation led to 48% OS in high-risk patients with 53% OS in high-risk patients with lung-only relapse on the NWTS-5 relapse protocol.239 The benefit of high-dose chemotherapy with autologous stem cell rescue (single or tandem transplants) is uncertain,242 although it has been shown that patients with residual disease do not do as well.243 Alternative agents, including topotecan244 and irinotecan,245 have shown some promising antitumor activity in phase 1/2 studies and require further evaluation in clinical trials.
Radiation Therapy
Successive NWTSG trials refined the dosages and indications to decrease radiation exposure while maintaining local control and control of metastatic pulmonary lesions (see Table 99.7). Patients with stage I and II favorable histology tumors do not require abdominal radiotherapy, whereas those with stage III favorable histology or stages I to III focal or diffuse anaplastic disease require adjuvant radiotherapy to the flank or abdomen. The tumor and renal beds with a 1- to 2-cm margin are treated in cases of positive resection margins, nodal involvement, or local spillage during surgery. Care must be taken to provide a uniform dosage to the vertebral column to limit the risk of scoliosis. Whole-abdominal radiotherapy is used for patients with preoperative rupture, diffuse spill during surgery, or when peritoneal metastasis is present. A dose of 10.8 Gy is sufficient for local control in stage III favorable histology patients if they also received chemotherapy with vincristine, dactinomycin, and doxorubicin.246 Most patients receive radiotherapy within 8 to 12 days of nephrectomy without compromise in local–regional control.247 Currently, it is recommended that radiotherapy start within 14 days of a nephrectomy.
Whole-lung irradiation (12 Gy) has been recommended for patients who present with pulmonary metastases, which has historically been defined by the presence of nodules on chest x-ray. However, CT now allows for the identification of much smaller lesions. Ehrlich et al.212 found that, in those patients whose pulmonary lesions were biopsied, only 82% of isolated lesions and 69% of multiple pulmonary lesions were actually tumor. Review of NWTS-4 and -5 identified patients with favorable histology and CT-only pulmonary lesions treated with standard three-drug therapy demonstrated no difference in EFS with or without the addition of radiation.248 For the SIOP 93-01 study, pulmonary radiation was successfully limited to those patients with unresectable, persistent nodules or high-risk primary tumor histology after receiving prenephrectomy chemotherapy for 6 weeks and postnephrectomy chemotherapy for 9 weeks.249 To develop a response-based approach to pulmonary radiation therapy, a recently closed COG trial evaluated the feasibility of eliminating radiotherapy in children whose chest CT scans demonstrated complete resolution of pulmonary nodules at week 6 of therapy. The results of this strategy are pending.
Retinoblastoma is a rare childhood cancer of the developing retina that often presents with leukocoria and is fatal if untreated. Early detection and judicious therapy based on laterality and stage of disease has resulted in survival rates exceeding 90% in the United States for localized (intraocular) disease.
Epidemiology
The incidence rate in the United States for the period 1975 to 1995 is estimated at 3.7 cases per million (about 300 new cases per year) with no racial predilection or significant difference between males and females.183 Worldwide, approximately 9,000 new cases are diagnosed each year.250 Over 85% of patients in the United States present with localized intraocular disease at diagnosis. Two-thirds of cases are diagnosed before 2 years of age and 95% before age 5 years. For unilateral disease, the median age at diagnosis is 2 years; for bilateral disease, the median age at diagnosis is less than 12 months. Approximately one-quarter of cases are bilateral and, therefore, have the heritable form of the disease (either sporadic or familial). In the remaining 75% of cases (unilateral), 10% to 15% of patients are found to have the heritable form of the disease. Parents and siblings of patients with retinoblastoma should undergo a thorough ophthalmoscopic examination while genetic testing is pending.
Biology and Genetics
Retinoblastoma is caused by the biallelic inactivation of the retinoblastoma gene, RB1, a tumor suppressor gene that is located on the long arm of chromosome 13 (13q14).251 Mutations in the RB1 gene are transmitted as a highly penetrant, autosomal-dominant trait.252 Patients with 13q- syndrome may have other associated abnormalities, such as distinct facial features, mental retardation, and growth failure.253 Additionally, these patients have more episodes of neutropenia and GI toxicity than other children with retinoblastoma when receiving systemic chemotherapy (Brennan, unpublished). Recently, rare cases of MYCN-amplified retinoblastoma without RB1 mutation have been reported.254 Despite the loss of a tumor suppressor gene, the retinoblastoma genome is relatively stable, with epigenetic dysregulation of cancer pathways, including upregulation of the proto-oncogene SYK, driving further tumorigenesis.255 Regardless of therapy, patients with heritable retinoblastoma are at an increased risk of developing secondary malignancies, particularly sarcomas, brain cancer, and melanoma256,257; those with familial, heritable retinoblastoma may be at an even higher risk.258 The cumulative incidence of secondary malignancies 50 years after diagnosis in a large US cohort was 36% for hereditary retinoblastoma survivors and 5.7% for nonhereditary survivors; however, the mortality was 25.5% for hereditary retinoblastoma survivors and 1.0% for nonhereditary survivors.259
Pathology
Retinoblastoma is composed of uniform small, round, or polygonal cells, which have a scanty, poorly staining cytoplasm. The sparse cytoplasm is located at one side of the cell, suggesting the appearance of an embryonal retinal cell. The nucleus is large and deeply staining, giving the characteristic small, round, blue cell appearance. Three types of cellular arrangements may be identified: the Homer-Wright rosette, the Flexner-Wintersteiner rosette, and the fleurette. Calcification and necrosis are often observed.260
Clinical Presentation
Leukocoria, an abnormal white reflection from the retina compared with the normal red reflex, is the most common presenting sign of retinoblastoma.261 Strabismus, conjunctival erythema, and decreased visual acuity are other common presenting complaints. Esotropia or exotropia may be present on examination, with decreased visual acuity as a result of involvement of the macula by the tumor or the presence of tumor cells and debris in the vitreous.262 The eye may be red and painful because of uveitis after spontaneous necrosis of a retinal tumor or from neovascular glaucoma.
Evaluation
The diagnosis of retinoblastoma is based on clinical history (including family history) and results of examination of both eyes under general anesthesia. Once confirmed, the staging evaluation of a child with retinoblastoma should include an ultrasound of the orbits and MRI of the brain and orbits.263,264 An MRI is useful to evaluate the extent of extraocular (orbital or pineal gland) disease. Although optic nerve enhancement may be concerning for the extraorbital spread of disease, pathology (enucleation) remains the gold standard.265 Retinoblastoma may metastasize to the central nervous system, bones, or bone marrow.266,267 A further evaluation for metastatic disease with lumbar puncture, bone marrow aspiration/biopsy, and bone scan is reserved for patients with involvement of extraretinal structures, including the orbit or optic nerve, or when symptoms, signs, pathology, or diagnostic imaging studies suggest the involvement of distant sites.268,269
Staging
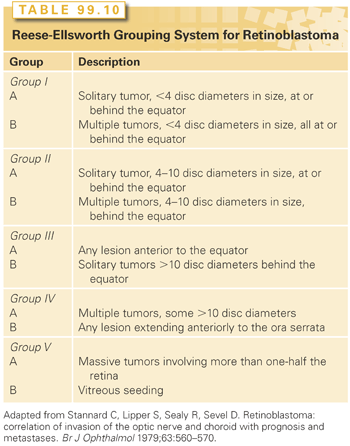
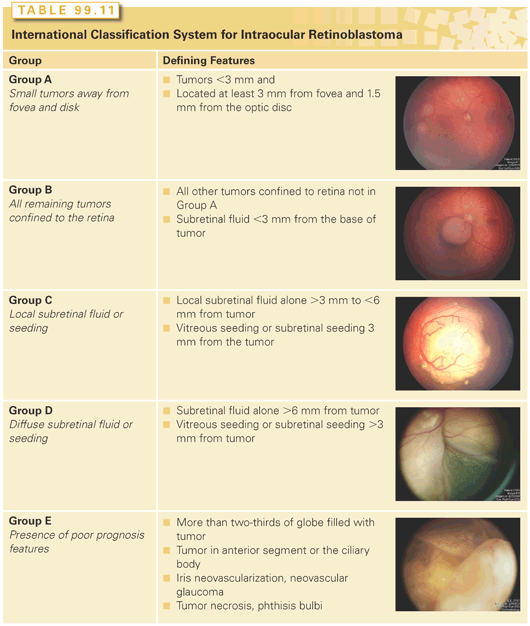
The patient is staged according to the burden of local and metastatic disease, whereas the eye is grouped according to the extent of intraocular disease present. Martin and Reese270 first proposed a staging system for patients with retinoblastoma in 1942. Two decades later, the Reese and Ellsworth271 grouping (R-E) attempted to predict the risk of enucleation with external-beam radiotherapy alone (Group Ia through Vb; Table 99.10). Although the R-E group (per eye) is still widely referenced today, the clinical value of utilizing a radiation-based system in an era of multimodal therapy that does not account for the presence of subretinal seeds and was developed before the introduction of indirect ophthalmoscopy is unclear. In 2001, the COG incorporated a new classification system (Group A through E) for grouping intraocular retinoblastoma that could be utilized among all institutions participating in clinical trials (Table 99.11). This classification system, which identifies the likelihood of treatment failure requiring enucleation or EBRT (group A = very low risk; group E = very high risk), was originally developed at Children’s Hospital Los Angeles and is now used internationally.272 The St. Jude Children’s Research Hospital developed a staging system that is defined by the extent of tumor involving the retina and globe, or by the presence of extrachoroidal disease.273 Another staging system developed by Chantada et al.274 provides a common international classification for patients with extraocular retinoblastoma.
Treatment
Patients with suspected retinoblastoma should be referred to a pediatric ophthalmologist with expertise in the management of intraocular tumors and a pediatric oncologist experienced in the treatment of retinoblastoma. Treatment aims to preserve life and useful vision and is based on laterality and the stage of disease. Protective eyewear is recommended for all patients. With the high percentage of survivors, treatment decisions must carefully weigh the functional outcome and potential long-term sequelae of local and systemic therapies.275
Enucleation
In view of the excellent response of this tumor to globe-sparing interventions, enucleation is considered in selective situations. Recently, more aggressive attempts at ocular salvage, regardless of visual potential, are being attempted (see the following). However, the most strongly considered indications for enucleation include (1) a unilateral retinoblastoma that completely fills the globe or that has damaged and disrupted the retina or vitreous so extensively that restoration of useful vision is not possible (and attempts at ocular salvage may place the patient at risk for metastatic disease); (2) a tumor that is present in the anterior chamber; (3) a painful glaucoma with a loss of vision after rubeosis iridis; (4) a progressive retinoblastoma disease unresponsive to all other forms of local therapy; and (5) cases with permanent vision loss in which extraocular tumor is suspected. The standard surgical technique is modified to allow excision of the longest possible segment of optic nerve in continuity with the globe.276 After removal, the globe and optic nerve are inspected for evidence of extraocular extension of the tumor. A hydroxyapatite implant is placed in the muscle funnel, which improves the cosmetic result and the promotion of normal development of the bony orbit.277
Local Therapies
When combined with chemoreduction, local therapies, including laser, cryotherapy, and radioactive plaque brachytherapy, consolidate treatment and improve disease control for patients with R-E group I through IV disease.278,279 Patients with R-E group V disease may require additional therapy, including external-beam radiation.280
Laser is useful for tumors ≤2.5 mm in thickness and ≤4.5 mm in diameter (<4 disc diameters).278 Like laser therapy, cryotherapy produces an avascular scar, but is more often utilized for tumors 2.5 mm in diameter and 1.0 mm thick located anterior to the equator and confined to the sensory retina.281 Radioactive plaque brachytherapy using iodine-125 radiation may be utilized for tumor consolidation, but is more commonly used in the setting of localized recurrence for tumor control or for salvage.282,283 Plaque brachytherapy does not appear to have the risk of secondary malignancies seen in external-beam radiation.283
Radiation Therapy
External-beam radiation therapy is indicated for patients with advanced disease that has not responded adequately to chemotherapy and local treatments but who has useful vision, or patients with extraorbital extension of disease, usually after enucleation. Patients requiring radiation receive doses between 40 and 45 Gy, depending on tumor size, patient age, and the presence of vitreous seeding.284 Modern radiotherapy uses megavoltage accelerators and CT- and MRI-based treatment planning to allow for sparing of the lens and bony orbits. Together with daily image guidance for field placement, these techniques may further reduce known radiotherapy-related late effects to surrounding structures.285 Proton therapy can potentially decrease dosage to the surrounding bony orbits by more than 60% and avoid significant dosage to the hypothalamus compared with intensity-modulated radiotherapy, with equivalent target coverage.286
The preservation of some useful vision in these patients is an obvious advantage of such a treatment approach, but the risk of secondary malignancies after external-beam radiation in young children is well defined.259,287 Kleinerman et al.288 published long-term results of 1,601 survivors of retinoblastoma and reported a 3.1-fold increase in the risk of secondary cancers for hereditary retinoblastoma survivors who received radiation therapy. Among hereditary retinoblastoma survivors, radiation therapy increased the cumulative incidence of second malignancies to 38% in patients who received radiation compared with 21% of hereditary patients without radiation. The authors also found the risk of osteosarcomas was higher in patients who received chemotherapy with radiation, compared with radiation alone.
After enucleation, local irradiation to include the orbit and the optic nerve up the chiasm is recommended for all patients with extension of retinoblastoma into the orbit. Presentation with exophthalmos, inability to retain the prosthesis, or a palpable mass through the eyelids suggests the presence of orbital extension of the tumor. The identification of an extraocular mass, histologic confirmation of tumor cells at the cut end of the optic nerve at the time of enucleation, or rupture of the globe during removal is associated with orbital contamination with the tumor.
Chemotherapy
The use of neoadjuvant chemotherapy in treating retinoblastoma was prompted by success in salvaging patients with recurrent extraocular disease.289,290 Since the 1990s, there have been a series of studies documenting promising rates of vision-sparing therapy without external-beam radiotherapy in intraocular disease.280,291,292 Most chemoreduction regimens are platinum based and utilize carboplatin over cisplatin (due to reduced ototoxicity). Other active agents include vincristine, etoposide, cyclophosphamide, doxorubicin, and topotecan.291,293–297 Cyclosporin A has also been used as a multidrug-resistant reversal agent.298 Despite the initial response, which is most pronounced in patients with R-E group I through IV tumors, chemotherapy alone rarely achieves durable disease control and is, therefore, paired with local therapies, as described previously. Patients with R-E group V disease, especially those with vitreous seeds, are at highest risk for chemoreduction failure and tumor recurrence/resistance.
Because tumor shrinkage may prevent enucleation, patients with bilateral disease are candidates for conservative treatment with chemoreduction. Indeed, in bilateral retinoblastoma cases, the assessment of which eye is most likely to be capable of functional vision is difficult until a response to chemotherapy is determined. Adjuvant chemotherapy is utilized after enucleation in patients who have high-risk histologic features, including invasion of the choroid (massive), anterior chamber, or ciliary body/iris, postlaminar optic nerve involvement with concomitant invasion of the choroid, or involvement of sclera or tumor present at the cut end of the optic nerve.295,299
Other recent areas of clinical investigation include locally delivered chemotherapeutic agents to increase drug concentration within the eye such as periocular chemotherapy (subconjunctival or subtenon injection),300 supraselective intra-arterial chemotherapy,301,302 and intravitreal chemotherapy.303 Because several reports have shown promising results, the techniques of intra-arterial and intravitreal chemotherapy will be prospectively evaluated in upcoming COG clinical trials to carefully evaluate the feasibility and toxicity of each regimen. At the same time, preclinical retinoblastoma research is providing novel therapies that target key pathways in retinoblastoma tumorigenesis.255,304,305 Pairing the right drug and delivery technique will translate into therapy that can save globes and vision while reducing the long-term effects of radiation and chemotherapy.
Metastatic Disease
Although most patients in the United States present with localized (intraocular) disease, 10% to 15% of patients will have evidence of extraocular spread at diagnosis. Patients with heritable disease (RB1 mutation) have a 3% to 9% risk of developing trilateral disease, or disease invading the pineal gland, although this may be lower in this era of improved MRI (tumor versus cyst of pineal gland) and systemic chemotherapy.264
Therapy for patients with metastatic disease requires a multimodal approach with intensive systemic chemotherapy, external beam radiation, and, for patients with distant systemic or central nervous system (CNS) metastatic disease, autologous bone marrow transplantation.289,306–308 Despite aggressive therapy, survival for patients with metastatic retinoblastoma outside of the CNS is 50%, with survival decreasing to <10% for those with CNS metastatic disease.
Late Effects and Long Term Follow-Up
Although patients treated for intraocular retinoblastomas have an excellent prognosis, those with germ-line RBI mutations remain at risk for secondary malignancies. Patients treated with chemotherapy are at risk for late effects, including secondary leukemia with exposure to etoposide309,310 and ototoxicity with exposure to carboplatin.311,312 There is also a growing awareness of the other long-term consequences that reach beyond the genetic and physical effects of therapy, such as health-related quality of life and neurocognitive and psychosocial outcomes.313,314 Further investigation through COG and other large retinoblastoma treatment centers is underway.
PEDIATRIC BONE SARCOMAS: OSTEOSARCOMA AND EWING SARCOMA
Osteosarcoma and Ewing sarcoma account for approximately 5% of all pediatric malignancies.183,315,316 Although the biologic properties of these tumors are distinct, their treatment principles are quite similar.317 Osteosarcoma has a bimodal age distribution, with the first peak in the 2nd decade of life and the second peak occurring in older adults over the age of 65 years. A comprehensive discussion of osteosarcoma is provided in Chapter 91. This section will focus on the main features of osteosarcoma in the younger population, including its unique molecular, genetic, and cytogenetic features.
Osteosarcoma
Epidemiology
Osteosarcoma is the most common primary, malignant bone tumor in children and adolescents, accounting for 4% of all childhood cancers. In the United States, approximately 400 new cases are diagnosed in those younger than 20 years of age.183 The peak age of incidence corresponds with the time of most rapid bone growth, 16 years in boys and 12 years in girls.318 The disease is slightly more prevalent in males and in African Americans.318
Biology and Molecular Genetics
The majority of osteosarcomas are sporadic. However, certain conditions, such as previous exposure to ionizing radiation319 and alkylating agents,320 predispose individuals to the development of osteosarcoma. Additionally, a large proportion of patients older than 40 years who develop osteosarcoma also have Paget disease of bone.321 Osteosarcoma is also associated with several cancer-predisposition syndromes, including hereditary retinoblastoma, Li-Fraumeni syndrome (LFS), and Rothmund-Thomson syndrome (RTS).
Patients with hereditary retinoblastoma have germ-line mutations in the RB1 gene and somatic mutations in retinal cells, resulting in retinoblastoma. The majority of secondary nonocular malignancies in these patients are sarcomas, and more than a third of these are osteosarcomas, half of which occur within a previously irradiated field.257,287,322 LFS is a familial cancer syndrome in which affected family members have a wide spectrum of cancers, including osteosarcoma.323 Many of these patients carry germ-line mutations in the p53 tumor suppressor gene.324,325 Screening of a large series of children with osteosarcoma showed that approximately 3% to 4% carried constitutional germ-line mutations in p53.326 RTS is an autosomal-recessive condition characterized by a distinctive rash (poikiloderma), small stature, skeletal anomalies, sparse hair, and increased risk for OS. In a cross-sectional study of 41 patients with RTS, 13 patients (30%) had osteosarcoma.327 Two-thirds of patients had constitutional mutations in the RECQL4 gene, and the presence of mutations correlated with osteosarcoma risk.328 Because these genetic conditions are known to predispose individuals to osteosarcoma, careful detailing of family history in a patient with newly diagnosed osteosarcoma is important to identify underlying genetic risk and for genetic counseling of family members.
In view of these genetic predisposition syndromes, it is not surprising that the RB1 and p53 genes are also frequently altered in sporadic osteosarcoma tumors. Approximately 70% of primary osteosarcoma tumors have alterations in the RB1 gene.329–331 Regulators of the RB1 pathway, including cyclin-dependent kinases 4 and 6, cyclin D1, and p16INK4a, are also altered in some cases of osteosarcoma.332–334 Inactivating mutations of the p53 gene occur in approximately 50% of all sporadic cancers. The overall frequency of p53 mutations in osteosarcoma ranges from 15% to 30% depending on the detection methods used.335 Other members of the p53 pathway, including p14ARF and MDM2, are altered in some cases of sporadic osteosarcoma.336,337 Unlike p53 and RB1, the RECQL4 gene has not been found to be mutated in cases of sporadic osteosarcoma.338
Clinical Presentation and Natural History
Most patients with osteosarcoma present with pain and soft-tissue swelling. Approximately 5% to 10% may present with a pathologic fracture of the affected bone.339,340 Systemic symptoms such as fever, weight loss, and malaise are generally absent. A physical examination usually demonstrates a firm, tender mass and restricted range of motion in the affected extremity. Laboratory evaluation results are usually normal except for elevated alkaline phosphatase (in approximately 40%),341 elevated lactate dehydrogenase (in approximately 30%),342 and elevated erythrocyte sedimentation rate, none of which are specific for osteosarcoma.
Osteosarcoma in the pediatric population preferentially involves the metaphyseal region of the long bones, in contrast to Ewing sarcoma, which typically arises in the diaphyseal region of the long bones. The primary site of disease in 80% of patients with osteosarcoma is an extremity, most commonly (in descending order) the distal femur, proximal tibia, and proximal humerus. Unlike Ewing sarcoma, osteosarcoma rarely affects the axial skeleton.343 Approximately 20% of patients with osteosarcoma present with clinically detectable metastatic disease, most frequently to the lungs and less often to bones.
Diagnostic and Staging Evaluation
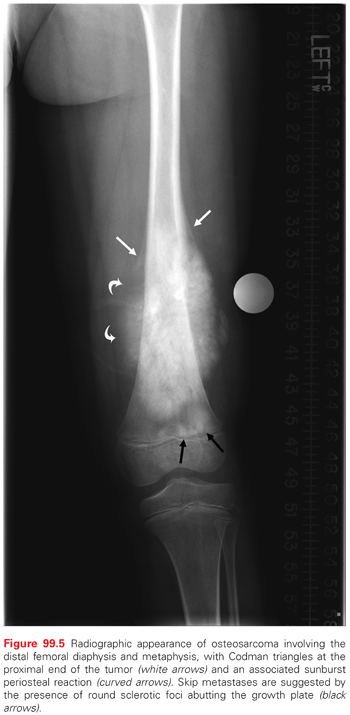
The initial evaluation of a suspected bone tumor involves obtaining a patient’s complete history and performing a physical examination. Radiographic studies allow for an assessment of the anatomic site, the extent of local invasion, and the pattern of extension.344 If the tumor involves an extremity, then plain radiographs should encompass both proximal and distal joint regions, and they should be taken in two planes. Characteristic findings suggestive of osteosarcoma on a radiograph include a mixed lytic and sclerotic appearance, periosteal new bone formation with lifting of the cortex and the formation of the Codman triangle, and ossification of the soft tissue in a radial or sunburst pattern (Fig. 99.5).343 Even if the plain radiographs are classic for osteosarcoma, further imaging of the primary tumor by MRI or CT is required to evaluate the extent of the tumor for surgery-planning purposes. The imaging should include the entire tumor-bearing bone in order to assess for skip metastases (i.e., isolated tumor foci within the same bone as the primary tumor). Although both CT and MRI are equally accurate for local staging of tumors, an MRI is preferable for evaluating soft-tissue extension and joint and marrow involvement.344
Because patients with metastatic disease at presentation have a significantly worse outcome than do patients with localized disease, a thorough search for sites of metastases is imperative. Staging workup for osteosarcoma includes a technetium-99 bone scan to evaluate the involvement of other bones and a chest CT to detect pulmonary metastases. Studies evaluating the utility of FDG PET/CT in osteosarcoma staging show that FDG PET/CT may be more sensitive and accurate than a bone scan in detecting bone metastases.345,346 Furthermore, the results of several studies suggest that changes in the metabolic tumor activity as assessed by PET/CT during therapy correlate with the percentage of tumor necrosis at the time of definitive surgery.347,348 A histologic confirmation is indicated in pulmonary lesion(s) that cannot be unequivocally defined as being a metastatic disease. Finding one or more pulmonary (or pleural) nodules of at least 1-cm diameter or three or more nodules of at least 0.5-cm diameter generally indicates definite pulmonary metastases and may not require a biopsy. Fewer or smaller lesions may or may not represent metastatic disease; therefore, confirmation by resection may be indicated.344 There are no laboratory studies that are diagnostic or prognostic for osteosarcoma.349 General laboratory tests such as a complete blood count; electrolyte counts including calcium, magnesium, and phosphorus; liver and renal function tests; and alkaline phosphatase and lactate dehydrogenase (LDH) measurements should be performed to obtain baseline values.349
Pathology
None of the radiographic features described previously are pathognomonic for osteosarcoma; therefore, a tissue biopsy is required to make a definitive diagnosis. The differential diagnosis for a bone lesion with aggressive features (e.g., the presence of a Codman triangle, associated soft-tissue mass, permeative appearance) on imaging studies includes Ewing sarcoma, lymphoma, and metastatic tumor. It may also occasionally include benign bone lesions, such as osteochondroma and giant cell tumor, and nonneoplastic conditions, such as osteomyelitis, eosinophilic granuloma, and aneurysmal bone cyst. The biopsy should be performed by an orthopedic surgeon experienced in the management of malignant bone tumors, ideally by the same surgeon who will perform the definitive surgery. Proper planning of the biopsy with careful consideration of the future definitive surgery is important so as not to jeopardize the subsequent treatment, particularly in the case of a limb-salvage procedure.350
Osteosarcoma is thought to be derived from primitive bone-forming mesenchymal cells.135 The histologic diagnosis is based on the presence of a malignant sarcomatous stroma associated with the production of tumor osteoid or immature bone.351 Several types of osteosarcomas have been identified based on histologic, clinical, and radiographic features including conventional osteosarcoma (80%), which is the type most frequently encountered in children and adolescents. Conventional osteosarcomas are further subdivided into osteoblastic (50% of conventional OS), fibroblastic (25%), and chondroblastic (25%) variants depending on the presence of the predominant type of matrix.352 The main feature that distinguishes these tumors from the malignant fibrosarcomas and chondrosarcomas, which also arise from primitive mesenchymal cells, is the production of osteoid.351
Other variants of osteosarcoma include parosteal, telangiectatic, small-cell, periosteal, low-grade central, and high-grade surface.353,354 All variants are considered to be high-grade tumors except for low-grade central and parosteal osteosarcomas, which are considered to be low-grade tumors, and periosteal osteosarcoma, which is thought to be intermediate grade.354,355 Small-cell osteosarcoma can be histologically confused with other small, round cell tumors, especially Ewing sarcoma.356 The tumors may stain positive for CD99 and may contain the Ewing sarcoma breakpoint region 1 (EWSR1) gene rearrangement.357 Two other distinct variants of osteosarcoma have been described, osteosarcoma of the jaw and extraosseous osteosarcoma. Osteosarcoma of the jaw tends to occur in older patients, has an indolent course, and is more often associated with local recurrences than with distant metastases. Extraosseous osteosarcoma is rarely encountered and usually occurs after exposure to radiation.358
In contrast to other pediatric sarcomas, osteosarcomas do not have any specific translocations or other molecular genetic abnormalities that can serve as diagnostic or tumor-specific markers of disease. Cytogenetically, osteosarcoma tumors have complex numerical and structural chromosomal abnormalities with significant cell-to-cell variation and heterogeneity, highlighting the complexity and instability of the genetic makeup of osteosarcoma.359–363
Treatment
The mainstays of therapy for osteosarcoma are surgery and chemotherapy. The outcome of patients with nonmetastatic osteosarcoma has improved dramatically during the past 3 to 4 decades from an EFS rate of 10% to 20% to one of 65% to 70%, mostly because of the addition of adjuvant chemotherapy as well as improvements in surgical and diagnostic imaging techniques.343,364–366 The care of patients with osteosarcoma requires a team approach involving oncologists, orthopedic surgeons, pathologists, oncology nurses, physical therapists, social workers, and child life specialists.
Chemotherapy
Localized Disease. Because of current combinations of surgery and chemotherapy, long-term disease-free survival and OS rates for osteosarcoma are greater than 60%. Based on the results of cooperative group trials over the past years (Table 99.12), the current standard three-drug chemotherapy regimen for patients with localized disease includes doxorubicin, cisplatin, and high-dose methotrexate (MAP). Patients receive neoadjuvant chemotherapy with these drugs for approximately 10 weeks and then undergo definitive surgery, which is followed by adjuvant chemotherapy for approximately 20 weeks. The exceptions to this therapy scheme in localized disease are low-grade central and parosteal osteosarcomas and some periosteal osteosarcomas without high-grade features; these variants are traditionally treated with surgery alone.354
The benefit of adjuvant chemotherapy in the treatment of osteosarcoma was clearly demonstrated in two prospective trials that randomly assigned patients to observation or chemotherapy following surgery.342,365,367 The concept of neoadjuvant or induction chemotherapy (i.e., chemotherapy given before definitive surgical resection) was prompted by the development of techniques for limb-sparing procedures and the need to control disease while the prosthesis was being constructed.368 In a prospective randomized trial by the Pediatric Oncology Group (POG) of neoadjuvant chemotherapy versus primary surgery followed by adjuvant chemotherapy369 outcomes were similar between the two groups, with 5-year EFS rates of approximately 61% and 69% (p = 0.8), respectively. Subsequent trials examining whether intensifying or adding other chemotherapy agents have not resulted in substantial improvements in OS for patients with osteosarcoma.370,371
The use of immunostimulatory agents in the treatment of osteosarcoma was tested in a joint POG and CCG randomized study (INT-0133).5 The aim of this study was to determine whether adding the macrophage-activating agent MTP-PE (muramyl tripeptide phosphatidylethanolamine; mifamurtide) to chemotherapy would enhance survival in patients with newly diagnosed osteosarcoma. The study also evaluated whether adding ifosfamide to the standard three-drug MAP regimen improved survival. Initial results suggested that there may be some modest benefit to patients treated with MTP-PE, and with additional follow-up of patients treated on this study show that adding MTP-PE had an improved 6-year OS rate from 70% to 78%. The drug is not available in the United States for clinical use.
The most recent study, an international trial (EURAMOS 1) for patients with resectable localized or metastatic osteosarcoma, has completed accrual. All patients were initially treated with standard three-drug induction with MAP. Those whose disease was classified as having a good response (>90% tumor necrosis) after induction chemotherapy continued with the same chemotherapy but were randomized to receive pegylated interferon α-2b, an immune-modulating cytokine that some Scandinavian studies have shown to have activity against osteosarcoma.372,373 Patients whose disease had poor histologic responses were randomized to receive high-dose ifosfamide and etoposide in addition to the standard three drugs. Preliminary results of the cohort of patients who experienced good responses to induction MAP chemotherapy showed no improvement in EFS rates of patients who received interferon α-2b versus those that received MAP alone; however, only 75% of patients who were randomized to receive interferon actually started the drug, and of those, only 55% completed all specified therapies.374
Metastatic Disease. The presence of metastatic disease at presentation is associated with a poor prognosis. In the absence of an available clinical trial, most patients with metastatic disease receive MAP chemotherapy with high-dose ifosfamide with or without etoposide.369,375,376
The ability to control all foci of macroscopic disease is essential in managing metastatic osteosarcoma. Patients with pulmonary metastatic disease have a survival rate of 30% to 50%, whereas patients with bone metastases have a worse prognosis.377,378 Similarly, patients with multifocal osteosarcoma have a dismal prognosis.
Multiple studies have demonstrated that the removal of all sites of metastatic or recurrent disease (e.g., pulmonary lesions), even after completion of chemotherapy, can result in long-term survival.379–381 In a large analysis of the Cooperative Osteosarcoma Study Group, which included more than 1,700 consecutively treated patients, the 10-year survival probability was 40% for metastatic patients who had all sites of metastatic disease resected.382 However, patients with extrapulmonary metastases (e.g., bone metastases) are less likely to be cured, particularly those with multifocal disease.383 Ongoing strategies to improve the outcome of patients with metastatic osteosarcoma include the use of MTP-PE,384 bisphosphonates,385,386 and molecularly targeted agents such as those against vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) receptors, mammalian target of rapamycin (mTOR), and Src kinase.387
Local Control. Osteosarcoma is relatively radioresistant; therefore, surgery alone is the mainstay of local control. The choice of limb salvage versus amputation for extremity tumors depends on the location and extent of the tumor, the ability to achieve good surgical margins, and proximity to the joints and neurovascular bundle. As long as the tumor is removed in its entirety with disease-free margins, then the type of surgery—limb salvage versus amputation—does not seem to influence outcome.388 On the basis of the results of studies assessing quality of life and functional measures, most patients who undergo limb salvage procedures report an improved quality of life and demonstrate better functional ability than do those who have amputations.389,390
Pelvic Tumors and Unresectable Disease. Patients with primary tumors of the axial skeleton generally have poor outcomes because surgery does not provide adequate local control. In some pelvic and most vertebral primary tumors, complete resection often is not possible. Most pelvic osteosarcomas can be treated by hemipelvectomy; however, more centrally located pelvic tumors, especially those involving the sacrum, are unresectable. Only a few pelvic osteosarcomas can be treated by limb-sparing resection (i.e., internal hemipelvectomy). Contraindications to resection are unusually large extraosseous extensions with sacral plexus or major vascular involvement. In general, these tumors cannot be resected with negative margins and are best treated by chemotherapy and radiotherapy.
Radiation Therapy. Historically, osteosarcoma has been considered to be relatively radioresistant; therefore, radiation therapy is generally not used as a definitive primary treatment of this disease. Radiation may be used in patients with microscopic positive margins of resection in doses of 55 to 68 Gy with local control rates ranging between 67% and 78%.391 Patients with gross residual or unresected disease have been treated to doses of 70 Gy or higher. Treatment volumes have consisted of the gross residual disease or tumor bed plus a 1- to 2-cm clinical margin. In the setting of metastatic or unresectable osteosarcoma, radiotherapy may also be of benefit by improving pain in up to 76% of cases.392 Despite the relative rarity of patients with osteosarcomas requiring radiation, its use can be of benefit in the management of both local and metastatic sites of disease.
Recurrent Disease. Outcomes for patients with local or distant relapse are poor.393–395 Effective therapy for these patients is challenging and is limited by the paucity of salvage chemotherapeutic agents active against this disease. Chemotherapy agents that have been used in the relapse setting include high-dose ifosfamide,396 gemcitabine and docetaxel,397–399 cyclophosphamide, and etoposide.400 More recently, targeted agents such as sorafenib have been used.401 Isolated pulmonary disease with a single unilateral lung lesion is more likely to result in long-term disease-free survival than is bilateral pulmonary disease or bony metastasis. A longer disease-free interval (>12 to 24 months) from initial therapy is associated with longer disease-free survival.394,402,403
Prognostic Factors
The most important prognostic factors for survival in patients with osteosarcoma are the presence of metastatic disease and the extent of tumor necrosis following induction chemotherapy, with more than 90% necrosis (i.e., grades 3 or 4) being favorable and less than 90% (i.e., grades 1 or 2) necrosis being less favorable.370,404–407 Other prognostic variables have been less definitive in predicting outcomes. Some of these factors include age, tumor size, and tumor location.382 Location is important because axial tumors, such as those of the pelvis, skull, or vertebrae, fare worse because of the difficulty in achieving a complete surgical resection with disease-free margins.
Ewing Sarcoma
Epidemiology
The term Ewing sarcoma was previously used to refer to the least-differentiated of a group of small, round cell tumors that share a recurring chromosomal translocation (discussed in detail in the following paragraphs). Over the years, this group of tumors became designated as the Ewing sarcoma family of tumors and includes Askin tumors (Ewing sarcoma arising in the chest wall) and the more histologically differentiated peripheral primitive neuroectodermal tumors. The 2013 World Health Organization classification proposed that the phrase Ewing sarcoma be used to refer to this family of tumors.408 We will follow this recommendation in this section.
Ewing sarcoma is the second most common primary bone tumor in pediatric patients after osteosarcoma, accounting for approximately 2% of childhood malignancies. About 200 cases of Ewing sarcoma are diagnosed in the United States per year.183 Most of these tumors arise in the 2nd decade of life. There is a slight male predominance, and African and Asian children are rarely affected by this cancer.183 Ewing sarcoma can arise in bone and, less commonly (about 30%), in the soft tissue (i.e., extraosseous) anywhere in the body.409
Biology and Molecular Genetics
The cell-of-origin of Ewing sarcoma is unknown but is presumed to arise from a mesenchymal stem cell.410–413 These tumors characteristically have recurrent chromosomal translocations that, in almost all cases, involve the EWSR1 gene on chromosome 22 and a member of the ETS family of transcription factors.414,415 Of Ewing sarcoma tumors, 85% harbor t(11;22) (q24;q12), resulting in the fusion of the EWSR1 and FLI1 genes. Another 10% to 15% of tumors are associated with t(21;22) (q22;q12), which generates the EWSR1-ERG fusion gene. Less commonly, EWS, or the related protein FUS, is fused to another ETS family transcription factor (i.e., ETV1, EIAF, FEV, or ETV4), resulting in the same Ewing phenotype.416
Heterogeneity in the EWSR1-FLI1 fusion gene is based on the location of the chromosomal breakpoint. The most common rearrangement, designated type 1, consists of the first seven exons of EWSR1 fused to exons six to nine of FLI1. This fusion gene accounts for over half of all cases. The type 2 rearrangement, accounting for about 25% of cases, fuses EWSR1 to exon 5 of FLI1. There is no prognostic significance based on the type of fusion protein.417,418 As a chimeric transcription factor, EWS-FLI1 is believed to regulate a number of critical downstream target genes that have been implicated in tumor biology, including members of the Sonic Hedgehog and GLI1 pathways.419,420
Evidence shows that directly inhibiting EWS-FLI1’s oncogenic activity by altering its interaction with critical binding partners such as RNA helicase A (RHA) significantly alters tumor biology. A small-molecule inhibitor that disrupts the EWS-FLI1/RHA interaction decreases xenograft growth and increases tumor cell apoptosis.421 Additional evidence suggests that several intracellular signal transduction pathways are significantly altered in these tumors, including insulin-like growth factor receptor 1 (IGF-1R) and the phosphatidylinositol-3 kinase (PI3K)–mTOR pathway.422 Drugs targeting these pathways are being explored as possible new therapeutic agents for Ewing sarcoma, including anti–IGF-1R antibodies and mTOR inhibitors.423–426
Clinical Presentation and Natural History
Patients with osseous Ewing sarcoma typically present with localized pain and swelling in the affected bone and may have other nonspecific symptoms such as fever, decreased appetite, and weight loss, which are usually seen in advanced disease.427,428 In contrast to osteosarcoma, Ewing sarcoma is equally distributed between extremity and axial sites. The lower extremity—primarily, the femur—is the most common site of disease, followed by the pelvis and the chest wall.429 Ewing sarcoma can also affect nonosseous structures, and this usually occurs in the soft tissues, but it can also arise in the GI tract, the kidneys, the adrenal gland, the lung, and other rare sites. The presenting symptom in these cases is site specific.
Metastatic disease is present in approximately 25% of patients at the initial diagnosis.430 The most frequent site of metastases is the lungs, followed by the bones, and bone marrow.429 Other sites of metastases, such as the lymph nodes, the liver, or the brain, are relatively rare unless in end-stage disease.
There is no specific blood or urine test to diagnose Ewing sarcoma. Abnormal laboratory findings at the time of diagnosis may include elevated LDH and alkaline phosphatase levels. LDH is useful as a gauge of tumor burden and usually falls with effective therapy and rises with disease recurrence.431
Diagnostic Staging Evaluation
An evaluation of suspected Ewing sarcoma includes a radiographic examination of the primary tumor site and documentation of the presence or absence of distant metastases. Plain radiographs and MRI or CT scans should be initially obtained to characterize and define the local extent of the primary tumor. An MRI provides the most precise definition of the extent of the tumor and its relation to nearby nerves and vessels. Lesions that originate in the long bones characteristically involve the diaphyses, with extension toward the metaphyses. On plain films, a lytic or mixed lytic–sclerotic lesion is usually identified in the bone. A multilamellar periosteal reaction (i.e., onion skin appearance [Fig. 99.6]) and lifting of the periosteum (i.e., Codman triangle), or less frequently, radiating bone spicules, may be present. The lesion is usually poorly marginated and has a permeative and destructive pattern.
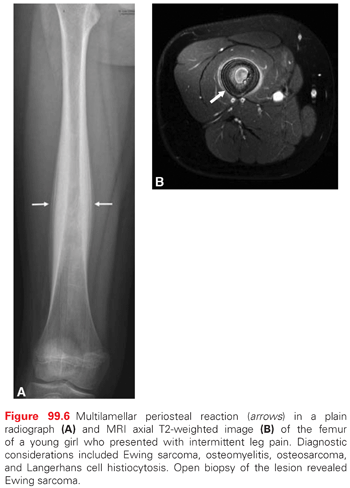
CT scans of the chest should be obtained to evaluate for pulmonary metastases. A biopsy of solitary pulmonary nodules should be strongly considered before classifying the disease as being metastatic. A technetium-99m whole-body radionuclide bone scan should be obtained to detect bone metastases. FDG PET/CT is highly sensitive in screening for bone metastases Ewing sarcoma432–434 and may be a useful predictor of outcome, similar to histologic response, after induction chemotherapy.435 Bilateral bone marrow sampling is required to complete the staging of all patients regardless of primary site or tumor size. Microscopically detectable bone marrow metastases occur in less than 10% of patients and are associated with a poor prognosis.436
Pathology
A biopsy of the tumor and an histopathologic examination are required for diagnosis. It is critical for the surgeon performing the diagnostic biopsy to place the incision appropriately to avoid complicating future resection. Under light microscopy, Ewing sarcoma falls under the category of small, round, blue cell tumors that includes neuroblastomas, rhabdomyosarcomas, lymphoblastic lymphomas, and, less commonly, histiocytosis and small-cell osteosarcomas. Ewing cells have a high nuclear-to-cytoplasmic ratio and appear homogenous, with uniform round nuclei that contain evenly distributed chromatin and little mitotic activity.415 The cytoplasm is typically scant and weakly eosinophilic; however, intracellular accumulation of glycogen may confer a positive periodic acid–Schiff test. Some tumors may demonstrate neuroectodermal differentiation. These tumors typically have Homer-Wright pseudorosettes on light microscopy and positive immunohistochemical staining for synaptophysin, neuron-specific enolase, S100, and CD57.415 More than 95% of Ewing sarcomas express CD99 (encoded by the MIC2 gene) on their cell membranes.429 Although CD99 staining of the cell membranes in a honeycomb pattern is suggestive of Ewing sarcoma, it is not pathognomonic and may be seen in a variety of tumors, including synovial sarcoma, mesenchymal chondrosarcoma, and B-cell and T-cell leukemia/lymphoma.437–440
The rapid identification of EWSR1 gene rearrangements by performing reverse transcription–polymerase chain reaction assays or fluorescence in situ hybridization assays on fresh, frozen, or paraffin-embedded specimens is useful for expeditiously discriminating between Ewing sarcoma and other morphologically similar small round cell tumors.414
Treatment
Chemotherapy. Ewing sarcoma is a very chemotherapy-sensitive tumor, and the outcome of patients with this disease has greatly improved as a result of intensifying systemic multiagent chemotherapy, particularly that of anthracycline and alkylating agents, as well as improvements in surgical and radiation techniques and supportive care (e.g., use of hematopoietic growth factors). Using current treatment strategies, over 70% of patients who present with localized disease are cured of their disease. However, the prognosis of patients with metastatic or recurrent disease remains poor, with a long-term survival rate of less than 30%.441
Localized Disease. Results of the major treatment trials for localized Ewing sarcoma are summarized in Table 99.13.411,442–454 Early cooperative group studies using adjuvant chemotherapy documented the efficacy of a four-drug regimen with vincristine (V), actinomycin D (A), cyclophosphamide (C), and doxorubicin (D) in combination with local control measures. By using these strategies, survival rates were improved from less than 20% to more than 40%.448,449
Related posts:




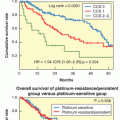
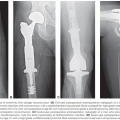
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


