PHARMACOGENOMICS OF TUMOR RESPONSE
Tumor response to chemotherapy is regulated by a complex, multigenic network of genes that encompasses inherent characteristics of the tumor, differentially activated pathways of cell signaling, proliferation and DNA repair, factors that control drug delivery to the tumor cells (e.g., metabolism, transport), and cell death. These may in turn be modulated by previously administered treatment or drug exposure, which may upregulate target proteins or activate alternative pathways of drug resistance. The polygenic nature of drug response implies that a better understanding of genotype–phenotype associations would require more than the usual single-gene pharmacogenetic strategies employed to date. However, there are instances where the genomic context of a single gene within a cancer will be of high impact for specific therapeutic agents (see Table 16.1).
Pathway Directed Anticancer Therapy
One of the earliest success stories illustrating pathway-driven therapeutics is with CML. The hallmark chromosomal abnormality of this disease is the translocation of chromosomes 9 and 22 that ultimately produces the fusion gene BCR-ABL. This discovery in 1960 eventually led to the development of the targeted tyrosine-kinase inhibitor (TKI) imatinib and its subsequent Food and Drug Administration (FDA) approval for treatment of CML in 2001.6 The International Randomized Study of Interferon and STI571 (IRIS) trial began enrollment in 2000 and compared imatinib with interferon and low-dose cytarabine, which was the previous standard of care for newly diagnosed patients with chronic-phase CML. All efficacy endpoints favored imatinib, including complete cytogenetic response of 76.2% with imatinib compared with 14.5% with interferon (p <0.001).7 Overall survival (OS) after 60 months of follow-up was 89% with imatinib.8 This example is just one of many where a once fatal disease can now be considered more akin to a chronic disease, requiring a daily medication and regular physician follow-up, similar to hypertension or diabetes. Drug development has also kept pace with these advances and now several other agents, including dasatinib, nilotinib, bosutinib, and ponatinib, have joined imatinib as treatment options for CML.
The idea of changing treatment focus from a disease-based model to a pathway-driven model is also evolving. Human epidermal growth factor receptor 2 (HER2) is a transmembrane receptor tyrosine kinase that is overexpressed or amplified in up to 25% of breast cancers. Trastuzumab is a humanized monoclonal antibody directed against HER2 and demonstrated improved response rates (RR) and time to disease progression in patients with metastatic HER2 positive breast cancer and improved disease-free survival (DFS) and OS in HER2-positive breast cancer patients treated with adjuvant trastuzumab.9 Several additional agents are now available to target the HER2 pathway and vary in their pharmacology and mechanism of action. Lapatinib is an oral TKI directed against HER2 and the epidermal growth factor receptor (EGFR), pertuzumab is a humanized monoclonal antibody that binds at a different location than trastuzumab and inhibits the dimerization and subsequent activation of HER2 signaling, and ado-trastuzumab emtansine is an antibody-drug conjugate that targets HER2-positive cells and then releases the cytotoxic antimitotic agent emtansine through liposomal degradation of the linking compound. All of these agents illustrate the progress and pharmacologic diversity of pathway-directed therapy and remain as standard of care options for HER2-positive breast cancer in either the adjuvant and/or metastatic settings.10 HER2 expression is not limited to breast cancer, however. Though less common, HER2 expression is seen in numerous solid tumors including bladder, gastric, prostate and non–small-cell lung cancer with varying degrees of incidence depending on the method of detection. Based on results from a large, open-label phase III randomized, international trial of 594 patients with gastric or gastroesophageal junction cancer expressing HER2 by either immunohistochemistry or gene amplification by fluorescence in situ hybridization, trastuzumab is also approved for treatment of metastatic gastric or gastroesophageal junction adenocarcinoma that expresses HER2. Patients randomized to chemotherapy in combination with trastuzumab had a median OS of 13.8 months compared with 11.1 months in the patients receiving chemotherapy alone (hazard ratio [HR], 0.74; 0.60 to 0.91, p = 0.0046).11 Numerous examples also support that pathway-directed therapy will cross the boundaries of disease sites and that tumor genetics will become one of the biggest determining factors for treatment.
Simple expression of the drug target does not always translate into desired clinical outcomes though. Cetuximab and panitumumab are monoclonal antibodies directed against EGFR; however, it was found that colorectal cancer (CRC) patients who did not have detectable EGFR still experienced responses to these agents similar in extent to EGFR-positive patients. Kirsten rat sarcoma viral oncogene (KRAS) is a downstream effector of the EGFR pathway. Ligand binding to EGFR on the cell surface activates pathway signaling through the KRAS-RAF-mitogen-activated protein kinase (MAPK) pathway, which is thought to control cell growth, differentiation, and apoptosis.12 Eventually it was found that CRC patients with a KRAS mutation did not derive benefit from cetuximab or panitumumab. The RR in CRC receiving either cetuximab or panitumumab who were KRAS wild type was 10% to 40% compared with near zero percent in those with KRAS mutations.13 This finding was the result of a retrospective analysis of small group of patients and was confirmed in large, prospective trials. Additionally, it underscores the importance of tissue collection for biomarker assessment in trials with novel therapeutics. A recent clinical trial genomic analysis suggests that mutations in NRAS may also have value in predicting the utility of EGFR antibody therapy in colorectal cancer. Although the predictive value of KRAS mutation status in colorectal cancer has been well established in clinical trials, the role of KRAS in lung cancer and other malignancies is less well elucidated. Lung cancers harboring KRAS mutations have been shown to have less clinical benefit from the EGFR-targeted erlotinib in some trials, although this has not consistently been the case across all trials. Additionally, lung cancer KRAS mutation status does not appear to reproducibly predict clinical benefit from the EGFR-targeted monoclonal antibodies, as is the case in colorectal cancer.14 Unlike the HER2 example discussed previously, the clinical application of some genetic mutations will differ between tissue of origin.
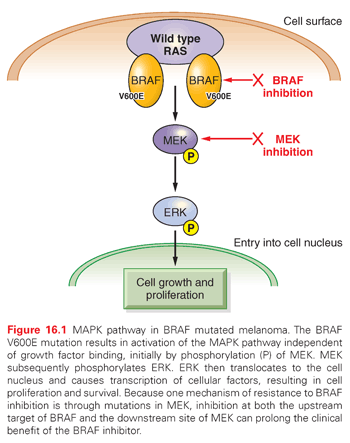
Deeper investigations and understandings of mutations driving oncogenic pathways can also elucidate mechanisms of resistance and practical therapeutic strategies for treatment and prevention. Approximately half of all cutaneous melanomas carry mutations in BRAF, with the most common being the V600E mutation. Vemurafenib is a TKI directed against mutated BRAF that demonstrated improvements in both progression-free survival (PFS) and OS when compared with the cytotoxic agent dacarbazine in previously untreated patients with metastatic melanoma carrying the BRAF V600E mutation. Vemurafenib demonstrated a 63% relative reduction in the risk of death compared with dacarbazine (p <0.001) along with a higher response rate (48% compared with 5% for dacarbazine).15 Based on these results, vemurafenib was the first BRAF targeted TKI approved by the FDA and was soon joined by dabrafenib. Although dramatic responses to these agents have been observed, relapse almost universally occurs after a median of 6 to 8 months. Activating BRAF mutations, like V600E, result in uncontrolled activity of the MAPK pathway through activation of the downstream kinase MEK, which when phosphorylated, subsequently activates extracellular signal-regulated kinase (ERK), which ultimately translocates to the cell nucleus, resulting in cell proliferation and survival (Fig. 16.1).16 An assessment of serial biopsies from patients treated with vemurafenib suggested numerous mechanisms for acquired resistance, including the appearance of secondary mutations in MEK.17 This finding supports the clinical rationale for using combination therapy with a BRAF and a MEK inhibitor. The combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) was assessed in 247 metastatic melanoma patients with BRAF V600 mutations compared with dabrafenib alone. Median PFS was 9.4 months in the combination group compared with 5.8 months in the patients who received single agent therapy (HR, 0.39; 0.25 to 0.62, p <0.001). A complete or partial response was also higher in the combination therapy group (76% compared with 54%, p = 0.03). The occurrence of cutaneous squamous cell carcinoma, a known side effect of single-agent BRAF inhibitor therapy due to paradoxical activation of RAF in nonmutated cells, was also decreased in the combination therapy group (7% compared with 19%, p = 0.09), further supporting the evidence of downstream inhibition.18 Although combination therapy does prolong the time to disease progression, resistance still occurs in patients through a variety of mechanisms. Utilization of sequential biopsies and a genetic assessment will help to inform rationale combination and sequential pathway-driven therapy trials that will ultimately aid in better understanding and mitigation of common mechanism of resistance.
Although advances in basic science and drug development have translated many oncogenic driver mutations across tumor types into pathway-directed therapy, this is not the case for the majority. There are numerous examples of functionally relevant recurrent driver mutations that affect protein targets that are not currently druggable. Regardless of malignancy, one of the most commonly mutated tumor suppressors is the protein p53. Mutations can result in p53 acquiring oncogenic functions that enable proliferation, invasion, metastasis, and cell survival as well as coordinating with different proteins, such as EGFR, to enhance or inhibit its effects. However, a clinical application of p53 mutation data or directly targeting p53 has been limited, to date.19 PIK3CA encodes a catalytic subunit of phophoinositol-3 kinase (PI3K), which includes four distinct subfamily kinases involved in regulating cell growth, motility, proliferation, and survival. Direct inhibitors of the kinase, as well as downstream targets, including AKT (protein kinase B [PKB]) and mammalian target of rapamycin (mTOR), are being assessed to target these mutations. Therapeutic challenges include understanding the complex signaling network germane to each cancer and the role of kinases in each subfamily.20 Both the examples of p53 and PI3K illustrate the challenge of translating the multitude of somatic mutations into applications of available therapeutic agents.
Application of Genomewide Gene Expression Profiling to Guide Therapy
Single gene approaches may not reflect the overall complexity of genetic regulation of chemotherapy responses. Genomic strategies using global gene expression data are able to provide a more complete picture of the tumor through disease classification.21 These strategies may identify subgroups of patients with early disease that need adjuvant chemotherapy, those who will not benefit from standard therapy, or help with the selection of chemotherapy from a menu of potentially active agents. Oncotype Dx is a 21-gene assay with 16 tumor-associated genes and 5 reference genes used to predict the risk of distant local recurrence in estrogen receptor (ER)-positive, HER2-negative patients with node-negative or select node-positive breast cancer. Additionally, the test also provides predictive information on which patients may benefit from the addition of chemotherapy to hormonal therapy alone. The test ultimately reports a recurrence score (RS) on a continuous scale from zero to 100. Patients with an RS <18 are considered low risk, with a 10-year distant recurrence rate (DRR) of 6.8% (95% confidence interval [CI], 4 to 9.6); RS scores of 18 to 30 are at intermediate risk, with a 10-year DRR of 14.3% (CI, 8.3 to 20.3); and RS scores ≥31 are at high risk, with a 10-year DRR of 30.5% (CI, 23.6 to 37.4).22 Additionally, high-risk patients have the largest benefit from the addition of chemotherapy to hormonal therapy (HR, 0.26; 0.13 to 0.53), whereas low-risk patients have little benefit from the addition of chemotherapy and could consider hormonal treatment alone (HR, 1.31; 0.46 to 3.78). Intermediate risk patients are harder to classify, and clinical trials are underway to further address treatment recommendations for this group of patients.23 These type of assays are also in development and in clinical trials for a variety of other solid tumor and hematologic malignancies.
Genetic-Guided Therapy Practical Issues in Somatic Analysis
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


