The study of pediatric cancer and rare hereditary cancer syndromes and associations has led to the identification of numerous cancer genes, including dominant oncogenes, DNA repair genes, and tumor suppressor genes. These genes are important not only in hereditary predisposition, but also in the normal growth, differentiation, and proliferation pathways of all cells. Alterations of these genes have been consistently found in numerous sporadic tumors of childhood and led to studies of their functional role in carcinogenesis. The numerous properties of transformed malignant cells in culture or in vivo can be explained by the complex abnormal interaction of numerous positive and negative growth-regulatory genes. Pediatric cancers offer unique models in which to study these pathways in that they are less likely to be disrupted by nongenetic factors. The embryonic ontogeny of many childhood cancers suggests that better understanding of the nature of the genetic events leading to these cancers will also augment the understanding of normal embryologic growth and development.
This chapter begins with an outline of tumor suppressor genes—the most frequently implicated class of cancer genes in childhood malignancy. This leads into a discussion of molecular features of retinoblastoma, the paradigm of cancer genetics, followed by an analysis of the molecular pathways associated with other common pediatric cancers. Evaluations of the importance of molecular alterations in familial cancers, as well as new approaches in molecular therapeutics are also addressed.
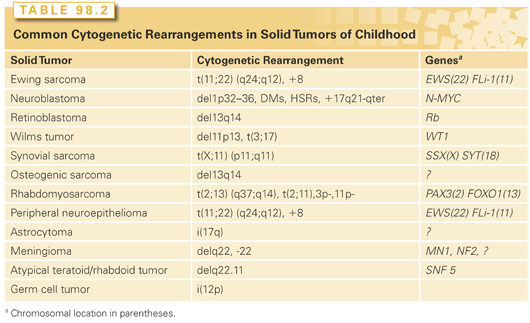
Faulty regulation of cellular growth and differentiation leads to neoplastic transformation and tumor initiation. Many inappropriately activated growth-potentiating genes, or oncogenes, have been identified through the study of RNA tumor viruses and the transforming effects of DNA isolated from malignant cells. However, activated dominant oncogenes themselves do not readily explain a variety of phenomena related to transformation and tumor formation. Among these is the suppression of tumorigenicity by the fusion of malignant cells with their normal counterparts. If these malignant cells carried an activated dominant oncogene, it would be expected that such a gene would initiate transformation of the normal cells, likely leading to either embryonic or fetal death. The observation is more readily explained by postulating the existence of a factor in the normal cell that acts to suppress growth of the fused malignant cells. Malignant cells commonly exhibit specific chromosomal deletions (Table 98.2). The best example of this occurs in retinoblastoma, a rare pediatric eye tumor in which a small region of the long arm of chromosome 13 is frequently missing. The presumed loss of genes in specific chromosomal regions argues strongly against the concept of a dominantly acting gene being implicated in the development of the tumor. Comparisons between the frequencies of familial tumors and their sporadic counterparts led Knudson3 to suggest that the familial forms of some tumors could be explained by constitutional mutations in growth-limiting genes. The resulting inactivation of these genes would facilitate cellular transformation.4 Such growth-limiting genes were termed tumor suppressor genes.
Whereas acquired alterations of dominant oncogenes most commonly occur in somatic cells, mutant tumor suppressor genes may be found either in germ cells or somatic cells. In the former, they may arise de novo or be transmitted from generation to generation within a family. The diversity of functions, cellular locations, and tissue-specific expression of the tumor suppressor genes suggest the existence of a complex, yet coordinated, cellular pathway that limits cell growth by linking nuclear processes with the intra- and extracytoplasmic environment. This discussion is limited to those genes for which pediatric tumors are frequently associated.
Retinoblastoma is the prototype cancer caused by mutations of a tumor suppressor gene. It is a malignant tumor of the retina that occurs in infants and young children, with an incidence of approximately 1:20,000.5 Approximately 40% of retinoblastoma cases are of the heritable form in which the child inherits one mutant allele at the retinoblastoma susceptibility locus (Rb1) through the germ line, and a somatic mutation in a single retinal cell causes loss of function of the remaining normal allele, leading to tumor formation. Tumors are often bilateral and multifocal. The disease is inherited as an autosomal-dominant trait, with a penetrance approaching 100%.6 The remaining 60% of retinoblastoma cases are sporadic (nonheritable), in which both Rb1 alleles in a single retinal cell are inactivated by somatic mutations. As one can imagine, such an event is rare, and these patients usually have only one tumor that presents itself later than in infants with the heritable form. Fifteen percent of unilateral retinoblastoma is heritable6 but by chance develops in only one eye. Survivors of heritable retinoblastoma have a several 100-fold increased risk of developing mesenchymal tumors such as osteogenic sarcoma, fibrosarcomas, and melanomas later in life.7 Several genetic mechanisms have been implicated in elimination of the second wild-type Rb1 allele in an evolving tumor. These include chromosomal duplication or nondisjunction, mitotic recombination, or gene conversion.8
The Rb1 gene maps to chromosome 13q14 and encodes a 105kD phosphoprotein.9,10 The second target gene that led to disease was actually the second copy of the Rb1 locus. Reduction to homozygosity of the mutant allele (or loss of heterozygosity [LOH] of the wild-type allele) would lead to the loss of functional Rb1 and account for tumor development. As well as being altered in retinoblastoma, Rb1 and its protein product are altered in osteosarcomas, small cell lung carcinomas, and bladder, breast, and prostate carcinomas.10,11 Rb1 plays a central role in the control of cell-cycle regulation, particularly in determining transition from G1 through S (the DNA synthesis) phase in virtually all cell types.
Although it is clear that Rb1 and its protein product play some role in growth regulation, the precise nature of this role remains obscure. In the developing retina, inactivation of the Rb1 gene is necessary but not sufficient for tumor formation.12 Retinoblastomas develop as a result of a complex interplay of aberrant expression of other cell-cycle control genes. In particular, a tumor surveillance pathway mediated by Arf, MDM2, MDMX, and p53 (see later discussion) is activated after a loss of Rb1 during development of the retina. Rb1-deficient retinoblasts undergo p53-mediated apoptosis and exit the cell cycle. Subsequently, amplification of the MDMX gene and increased expression of MDMX protein are strongly selected for during tumor progression as a mechanism to suppress the p53 response in Rb1-deficient retinal cells.13 Not only do these observations provide a provocative biologic mechanism for tumor formation in retinoblastoma, but it also offers potential molecular targets for novel therapeutic approaches to this tumor.14,15 Some Rb1 mutations appear to lead to an attenuated form of the disease, an observation that highlights the variable penetrance in families.16,17 Outside the retina, Rb1 inactivation is often a rate-limiting step in tumorigenesis generated by multiple genetic events. The molecular characteristics and potential functional activities of Rb1 are outlined in detail elsewhere in this volume.
The patterns of inheritance and presentation of retinoblastoma have been well described and the responsible gene has been identified. The basic mechanisms by which the gene is inactivated are understood, and provocative evidence indicates that the intricate functional interactions of pRB with its binding partners and other cell cycle targets will provide targets for development of novel small molecule therapies.
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors found in the gastrointestinal (GI) tract, and occur in both adults and children. Strikingly, unlike most adult GIST tumors that harbor either KIT or platelet-derived growth factor receptor A (PDGFRA) mutations and are susceptible to tyrosine kinase (TK) inhibitors directed against these kinases, 85% of pediatric GIST tumors do not harbor such mutations, and not surprisingly are much less responsive to TK inhibitors. Thus, it appears that a histologically similar tumor in a pediatric population is remarkably biologically distinct from the same tumor in the adult population. It was recently demonstrated that the pediatric-type GIST tumor is characterized by metabolic derangement of the mitochondrial enzyme, succinate dehydrogenase (SDH).18 Many of these patients have been found to carry germ-line mutations of SDH subunits B, C, or D (SDHx) mutations. These germ-line mutations were first described in Carney-Stratakis syndrome, an inherited predisposition to GIST and paragangliomas.19 It is noteworthy that these SDH-deficient pediatric predominant GISTs have a marked predilection for gastric location and are more frequent in the female population.20
Further characterization of molecular differences between the typical adult GIST, KIT, or PDGFRA activated tumors and SDH-deficient, pediatric-type GIST tumors revealed marked epigenetic distinctions between these two types of GISTs. Specifically, the SDH-deficient tumors were found to have marked global hypermethylation compared to the TK-mutant GISTs. Of specific note was enrichment for hypermethylation within DNase hypersensitivity sites.21
Thus it is clear that most pediatric GISTs are driven by a distinct mechanism related to SDH-deficient function compared to the adult GISTs, which are driven by the mutation activation of KIT or PDGFRA kinases. Furthermore, many of these tumors are associated with germ-line mutations of SDHx, and may or may not be associated with paragangliomas. The finding of global hypermethylation in the pediatric-type GISTs was associated with very stable genomes, suggesting these tumors are epigenetically driven tumors.21 Overall, these data clearly suggest that despite histologically similar tumors, the GISTs that occur in the younger population have a distinct behavior that will require management that is quite different than the management of adult, TK-mutating GISTs.
The neurofibromatoses comprise two similar entities. Neurofibromatosis type 1 (NF1) is one of the most common autosomal-dominantly inherited disorders, affecting about 1 in 3,500 people,22 half of which arise from new spontaneous mutations. Carriers of mutant NF1 are predisposed to a variety of tumors, including Schwann cell–derived tumors (neurofibromas and malignant peripheral nerve sheath tumors, gliomas including optic nerve gliomas and malignant gliomas, and pheochromocytomas).23,24 Occurring with less frequency are leukemias, osteosarcomas, rhabdomyosarcomas, GISTs, and Wilms tumors.
Using a standard linkage analysis, the neurofibromatosis type 1 (NF1) gene was mapped to chromosomal band 17q11, and subsequently cloned.25,26 The NF1 gene is unusual in that it contains three embedded genes, OMGP, EV12A, and EV12B, in a single intron.27 This gene encodes a 2818 amino acid protein, termed neurofibromin, that is ubiquitously expressed. One region of the gene shows extensive structural homology to the GTPase activating domain of mammalian GTP-ase–activating protein (GAP) proteins: loss of the protein’s activity results in failure of hydrolysis of GTP to guanosine diphosphate (GDP) by the ras oncoprotein. Loss of neurofibromin function usually results from mutations in one allele of the gene leading to premature truncation of the protein, followed by absence or mutations of the second allele in tumors. This loss of function is thought to lead to elevated levels of the GTP-bound RAS protein that transduces signals for cell division. Downstream modulators of RAS activation in NF1-associated tumors include activation of mammalian target of rapamycin (mTOR) and mitogen activated protein kinase kinase (MEK).28,29 Thus, although targeting RAS remains elusive, mTOR and MEK inhibitors are currently either available or are being tested in the clinic. Neurofibromatosis type 2 (NF2) is much less frequent than NF1, occurring in only 1 in 1 million persons. Although it is also inherited as an autosomal-dominant disorder with high penetrance, the new mutation rate in NF2 is low.30 It is clinically characterized by bilateral vestibular schwannomas, spinal nerve root tumors, meningiomas, and ependymomas.
The NF2 locus was mapped to chromosome 22, band q12,31 and its 69-kDa encoded protein, termed merlin, has been shown to be expressed in various tissues, including the brain, although not as ubiquitously as NF1.32,33 The mechanism of tumor formation in NF2 appears to be in concordance with the Knudson two-hit model, although the mechanism of action of the NF2 protein has not yet been elucidated. Merlin is a member of the Band 4.1 family of proteins that link cell surface proteins to the cytoskeleton.34 Merlin has been shown to inhibit the number of signaling pathways critical for normal growth including mTOR, Rac1, and Hippo/YAP, with loss of Merlin leading to activation of these pathways, again suggesting potential therapies aimed at inhibition of these pathways.34
Nonrandom chromosomal abnormalities are observed in more than 75% of neuroblastomas.35 The most common of these is deletion or rearrangement of the short arm of chromosome 1, although loss, gain, and rearrangements of chromosomes 10, 11, 14, 17, and 19 have also been reported. The allelic losses indicate loss of function of as yet unknown tumor suppressor genes in these regions. It is believed that a tumor suppressor gene, such as CHD5 and the kinesin KIF1Bbeta that lies on band p36 of chromosome 1 is critically important in the pathogenesis and aggressive nature of neuroblastoma.36,37 It has been shown that the loss of chromosome 1p is a strong prognostic factor in patients with neuroblastoma, independent of age and stage.38 Although it is as yet unclear which gene(s) in this region may be directly implicated in neuroblastoma development, aberrant expression of one candidate—p73—which is a member of the p53 tumor suppressor family, has been suggested to play a role in the neuroblastoma cell growth as well as chemotherapy resistance.39 p73 gives rise to multiple functionally distinct protein isoforms as a result of alternative promoter utilization and alternative mRNA splicing.40,41 Alternative splicing of the p73 mRNA results in more than seven protein isoforms that differ in the coding sequences of the COOH terminus (TA-p73 α, β, γ, δ, ε, ζ, η).
In addition to these COOH-terminal splice forms, three additional forms, Np73α, ΔNp73β, and ΔNp73γ, are transcribed from an alternative promoter located in intron 3. Higher levels of ΔNp73 are associated with an overall worse clinical prognosis, presumably because of the antiapoptotic properties of ΔNp73 and its ability to inactivate both TAp73 and p53.42,43
Two other unique cytogenetic rearrangements are highly characteristic of neuroblastomas.44 These structures, homogeneous staining regions (HSR) and double-minute chromosomes (DM), contain regions of gene amplification. The N-myc gene, an oncogene with considerable homology to the cellular protooncogene c-myc, is amplified within HSR and DM. Virtually all neuroblastoma tumor cell lines demonstrate amplified and highly expressed N-myc,45 and N-myc amplification is thought to be associated with rapid tumor progression. Expression of N-myc is increased in undifferentiated tumor cells compared with much lower (or single-copy) levels in more differentiated cells (ganglioneuroblastomas and ganglioneuromas). N-myc expression is diminished in association with the in vitro differentiation of neuroblastoma cell lines.46 This observation formed the basis for current therapeutic trials demonstrating a survival advantage to patients treated with cis-retinoic acid.47 Furthermore, a close correlation exists between N-myc amplification and advanced clinical stage.48
Neuroblastoma cells that express the high-affinity nerve growth factor receptor trkA49 can be terminally differentiated by nerve growth factor and may demonstrate morphologic changes typical of ganglionic differentiation. Tumors showing ganglionic differentiation and trk gene activation have a favorable prognosis.49 In contrast, trkB receptor expression is associated with poor-prognosis tumors and appears to mediate resistance to chemotherapy.50,51 Resistance to multidrug chemotherapeutic regimens (i.e., multidrug resistance) is characteristic of aggressive, poorly responsive N-myc–amplified neuroblastomas. It is interesting to note that expression of the multidrug resistance–associated protein, found to confer multidrug resistance in vitro, is increased in neuroblastomas with N-myc amplification, decreased after differentiation of tumor cells in vitro, and associated with poor outcome independent of N-myc amplification.52 Gain of chromosome segment 17q21-qter, in which BICR5, which encodes Survivin (a member of the apoptosis inhibiting proteins NM23 and PMMID) has been posited as a neuroblastoma-associated candidate gene, has been shown to be the most powerful prognostic factor yet.53
A small subset of neuroblastomas is inherited in an autosomal-dominant fashion. Until recently, the only gene definitively associated with neuroblastoma risk was PHOX2B, also linked to central apnea.54 De novo or inherited missense mutations in the TK domain of the anaplastic lymphoma kinase (ALK) gene on chromosome 2p23 have been observed in the majority of hereditary neuroblastoma families, as well as in somatic tumor cells.55–58 Current phase I/II clinical trials with ALK inhibitors substantiate the value of such target identification for novel therapies.59 The role of other molecular alterations in neuroblastoma continues to be elucidated. In addition to chromosomal loss on chromosome 1p36, unbalanced LOH at 11q23 is independently associated with decreased event-free survival. Alterations at 11q23 occur in almost one-third of neuroblastomas, being most commonly associated with stage 4 disease and age at diagnosis greater than 2.5 years. Both 1p36 LOH and 11q23 LOH were independently associated with decreased progression-free survival in patients with low- and intermediate-risk disease.60
Yet another valuable biologic marker of clinical significance is telomerase expression and telomere length. In particular, short telomere length is predictive of favorable prognosis, irrespective of disease stage, whereas long or unchanged telomeres are predictive of poor outcome.61,62 Telomerase expression, as measured by human telomerase reverse transcriptase (hTERT), has been shown to be negative in good-risk neuroblastomas, although it is high in tumors with unfavorable histology.62 The combined use of these markers—chromosomes 1p and 11q, N-Myc amplification, trkA, and telomerase expression—as prognostic indicators provides a powerful armamentarium with which to develop rational stratified treatment programs for neuroblastomas.
EWING SARCOMA FAMILY OF TUMORS
Ewing sarcoma (ES) is one of the first examples in which the application of molecular diagnostics led to improved tumor classification. ES was first described by James Ewing63 as a bone tumor characterized by small, blue, round cells and minimal mitotic activity. Turc-Carel et al.64 identified a recurring reciprocal t(11;22) chromosomal translocation in these tumors in 1983. Investigators subsequently demonstrated a cytogenetically identical t(11;22) in adult neuroblastoma or peripheral primitive neuroectodermal tumor (pPNET), so named because of its histologic similarity to neuroblastoma.65 Based on the presence of the identical translocation, it was hypothesized that pPNET was related to ES. This translocation breakpoint has been molecularly characterized as an in-frame fusion between a new ES gene, Ewing’s sarcoma breakpoint region 1 (EWS), on chromosome 22 and an ETS transcription family member, friend leukemia virus integration 1 (FLI-1), on chromosome 22.66–68
In addition to this fusion transcript being identified in pPNET, other variants, notably the chest-wall Askin tumor and soft tissue ES—previously treated as an RMS because of its location in soft tissue—were also shown to bear the identical fusion transcript. In total, five translocations also have been identified, invariably fusing the EWS gene to an ETS family member.69–72 More than 90% of the ES family of tumors (ESFT) carry the EWS-ETS fusion gene, and a search for EWS-ETS by either reverse transcriptase-polymerase chain reaction or fluorescence in situ hybridization should be considered standard practice in the diagnostic evaluation of suspected ESFTs. Interestingly, although it was suggested that the specific fusion protein expressed in ESFT has prognostic significance,73 several prospective studies in the United States and Europe demonstrated no prognostic impact.74,75 The nature of the novel fusion transcription factor and its downstream targets is currently under intense investigation. One target of the EWS-ETS fusion is repression of the transforming growth factor-β type II receptor,76 a putative tumor suppressor gene.
Expression profiling analysis has also revealed that p53 is transcriptionally upregulated by the EWS-ETS fusion gene.77 This is of particular interest because it is now known that expression of EWS-ETS can lead to apoptosis, and that additional alterations such as loss of p53 or p16 signaling, or both, appear to be necessary components of EWS-ETS–induced transformation.78 Investigators have now taken advantage of RNA interference technology to inhibit EWS–FLI-1 in Ewing cell lines to identify genes regulated by the fusion in the proper context. Using this approach, NKX2.2 and NR0B1 have been found to be a target gene of EWS–FLI-1 that is necessary for oncogenic transformation.79,80 Recent findings suggest that GGAA microsatellites might mark genes that are upregulated by EWS–FLI-1 binding.81
The two major histologic subtypes of RMS, embryonal and alveolar, have both unique histologic appearance as well as distinctive molecular genetic abnormalities, while sharing a common myogenic lineage. Embryonal tumors comprise two-thirds of all RMS, and are histologically characterized by a stroma-rich spindle-cell appearance. Alveolar tumors comprise about one-third of RMS, and are histologically characterized by densely packed small round cells often lining a septation reminiscent of a pulmonary alveolus, giving rise to its name. Both histologic subtypes express muscle-specific proteins, including α-actin, myosin, desmin, and MyoD,82–84 and they virtually always express high levels of insulin-like growth factor 2 (IGF2).85,86
At the molecular level, embryonal tumors are characterized by LOH at the 11p15 locus, which is of particular interest because this region harbors the IGF2 gene.87,88 The LOH at 11p15 occurs by loss of maternal and duplication of paternal chromosomal material.89 Although LOH is normally associated with loss of tumor suppressor gene activity, in this instance, LOH with paternal duplication may result in activation of IGF2. This occurs because IGF2 is now known to be normally imprinted; that is, this gene is normally transcriptionally silent at the maternal allele, with only the paternal allele being transcriptionally active.90,91 Thus, LOH with paternal duplication potentially leads to a twofold gene-dosage effect of the IGF2 locus. Furthermore, in alveolar tumors where LOH does not occur, the normally imprinted maternal allele has been shown to be reexpressed.92,93 Thus, LOH and loss of imprinting (LOI) may in this case lead to the same functional result—namely, biallelic expression of the normally monoallelically expressed IGF2. However, loss of an as yet unidentified tumor suppressor activity due to LOH also remains a possibility.
Alveolar RMS is characterized by a t(2;13)(q35;q14) chromosomal translocation.94 Molecular cloning of this translocation has identified the generation of a fusion transcription factor, fusing the 5′ DNA-binding region of PAX-3 on chromosome 2 to the 3′ transactivation domain region of FOXO1 gene on chromosome 13.95,96 A variant t(1;13)(q36;q14) has been identified in a small number of alveolar RMS tumors that fuses the 5′ DNA-binding region of the PAX-7 gene on chromosome 1 with the identical 3′ transactivation domain of the FOXO1 gene.97 Fluorescence in situ hybridization (FISH) or reverse transcription polymerase chain reaction (RT-PCR) can be used to identify these PAX3/7-FOXO1 fusions in approximately 90% of tumors, and are diagnostic of alveolar rhabdomyosarcoma (ARMS). As noted previously, the fusion protein generated by the translocation leads to a novel transcription factor.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


