Solid and Trabecular Lesions
PAPILLARY CARCINOMA
Solid Variant
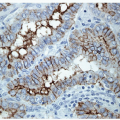
The solid variant of papillary carcinoma is characterized by solid nests of epithelial cells that have well-defined borders but fail to form follicles and lack colloid storage [1]. The tumor cells remain well differentiated and exhibit the nuclear features of papillary carcinoma (Fig. 11.1, e-Fig. 11.1). Solid variant papillary carcinoma must be distinguished from insular carcinoma, a form of poorly differentiated thyroid carcinoma that is composed of smaller, less well-differentiated cells and exhibits tumor cell necrosis (Fig. 11.2, e-Fig. 11.2). Although somewhat controversial, it appears that this solid variant of papillary carcinoma is associated with more aggressive behavior than classical papillary thyroid carcinoma [2, 3].
The fine needle aspiration (FNA) appearance of the solid variant is not necessarily recognized as a distinct entity but tends to show a clean background with only scant or no colloid and marked cellularity. The follicular epithelial cells are arranged in cohesive clusters with nuclear overlapping and crowding [4, 5]; a dyshesive single-cell pattern may be seen if an infiltrative lesion is biopsied [6]. The nuclear changes of papillary carcinoma are identified with prominent grooves and micronucleoli and occasional intranuclear inclusions.
The histologic diagnosis can be confirmed by the identification of specific markers. Solid lesions should be verified as of thyroid follicular differentiation by showing immunoreactivity for TTF-1 and thyroglobulin. These tumors stain for HBME-1 and galectin-3 [7] and should not show mitoses or necrosis, although the latter may be seen focally at the site of previous biopsy. Solid variant papillary carcinomas are known to harbor ret/PTC rearrangements, as shown particularly in children exposed to radiation after the Chernobyl disaster [2, 8]. Although the classical activating BRAF mutations are not found in this variant, the in-frame VK600-1E deletion (BRAFVK600-1E) has been detected in solid variant papillary thyroid carcinomas [4, 9, 10]. The distinction of this lesion from poorly differentiated “insular” carcinoma may require additional immunohistochemical stains (see below).
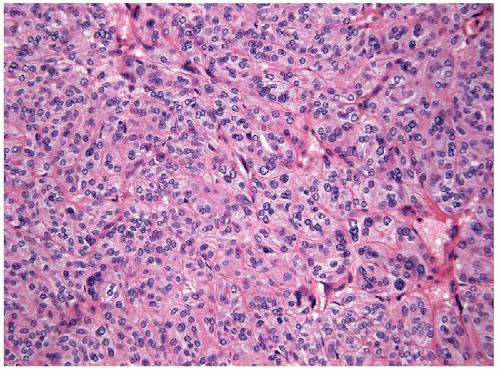 FIGURE 11.1 The solid variant of papillary carcinoma lacks a follicular architecture and colloid production but remains well differentiated with the classical nuclear features of papillary carcinoma evident (hematoxylin and eosin stain). |
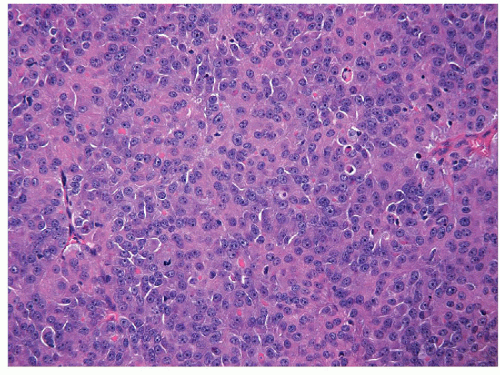 FIGURE 11.2 Insular carcinoma. Superficially, the solid variant of papillary carcinoma may mimic insular carcinoma. However, as seen in this image, insular carcinoma is characterized by less well-differentiated follicular epithelial cells, lacking the nuclear features of papillary carcinoma and often betraying their aggressive nature with foci of necrosis and readily identified mitoses (hematoxylin and eosin stain). |
Hyalinizing Trabecular Variant
An unusual tumor known as the hyalinizing trabecular tumor (HTT) was originally described by pioneers such as Zipkin in 1905 [11], Masson in 1922 [12], and Ward et al. in 1982 [13]. The terminology “hyalinizing trabecular adenoma” (HTA) was defined by Carney et al. in 1987 [14]. This lesion has also been designated “paraganglioma-like adenoma of thyroid” (PLAT) by Bronner et al. [15] because of its unusual histologic pattern that resembles a neuroendocrine tumor. Since the original descriptions, a malignant counterpart, hyalinizing trabecular carcinoma (HTC), has been described [16, 17, 18], and the designation of these as HTT was proposed [1].
HTTs share many features with papillary carcinoma. Both lesions are of thyroid follicular epithelial origin and therefore both express thyroglobulin. HTTs have been associated with Hashimoto’s thyroiditis and are reported in patients who have a history of neck irradiation [19]. HTT can coexist with papillary carcinoma [20]. Indeed, HTTs exhibit papillary carcinoma-like histologic features such as psammoma body formation and characteristic nuclear changes including elongation and irregularity with grooves and pseudoinclusions, and nuclear hypochromasia [14]. Based on these observations, as well as molecular genetic alterations, specifically ret/PTC rearrangements, that are shared by both entities [21, 22], many experts consider these to be variants of papillary thyroid carcinoma [1, 23, 24].
These lesions are generally well-delineated tumors characterized architecturally by solid trabecular and nesting architecture (Figs. 11.3 and 11.4, e-Fig. 11.3). The tumor cells are elongated with abundant pale
eosinophilic cytoplasm and scattered “yellow bodies” [14, 15, 19, 25]. There is perivascular hyaline fibrosis, and the cytoplasmic hyaline is usually identified as cytoplasmic filaments of cytokeratin. Most importantly, the tumor cells harbor large clear nuclei with irregular and elongated contours, grooves, and inclusions as well as micronucleoli, features of papillary carcinoma (Figs. 11.5, 11.6, 11.7, e-Fig. 11.4).
eosinophilic cytoplasm and scattered “yellow bodies” [14, 15, 19, 25]. There is perivascular hyaline fibrosis, and the cytoplasmic hyaline is usually identified as cytoplasmic filaments of cytokeratin. Most importantly, the tumor cells harbor large clear nuclei with irregular and elongated contours, grooves, and inclusions as well as micronucleoli, features of papillary carcinoma (Figs. 11.5, 11.6, 11.7, e-Fig. 11.4).
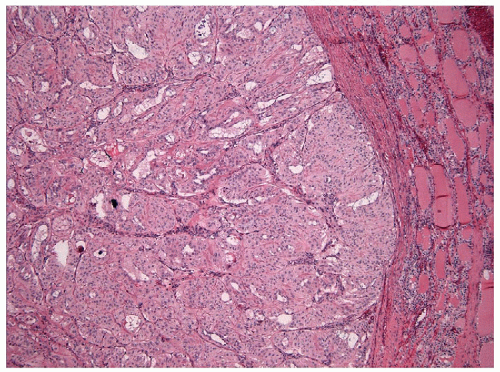 FIGURE 11.3 Hyalinizing trabecular tumors of the thyroid are well demarcated from the surrounding thyroid parenchyma, which may be appreciated grossly as well as microscopically (hematoxylin and eosin stain). |
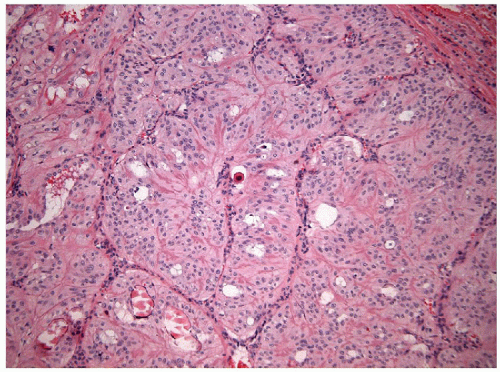 FIGURE 11.4 Hyalinizing trabecular tumors are composed of solid trabeculae, cords, and nests of cells with interposed hyalinized stroma (hematoxylin and eosin stain). |
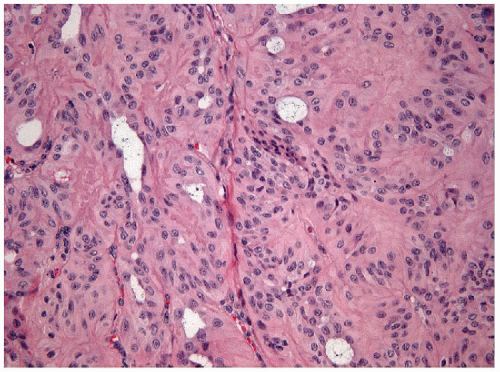 FIGURE 11.5 Hyalinizing trabecular tumor. The epithelial cells of hyalinizing trabecular tumors are elongated with abundant eosinophilic cytoplasm and often show an organoid arrangement around the hyalinized stromal tissue (hematoxylin and eosin stain). |
 FIGURE 11.6 Hyalinizing trabecular tumors display all of the classical nuclear features of papillary carcinoma and usually to an exuberant degree. In addition, cytoplasmic inclusions containing a central body surrounded by a halo are seen in many cases (arrows) (hematoxylin and eosin stain). |
The morphologic key to the recognition of these lesions on FNA is the identification of the aggregates of the hyalinized stroma with radially oriented, elongated/spindle epithelial cells with abundant cytoplasm (Figs. 11.8 and 11.9, e-Figs. 11.5, 11.6, 11.7). The cytoplasm is often somewhat
filamentous, and cell borders are indistinct. Romanowsky staining highlights the metachromatic hyaline material, perinucleolar clearing, and cytoplasmic bodies. Intranuclear cytoplasmic inclusions, nuclear grooves, and nuclear overlapping are the rule and well visualized on Papanicolaou staining [26, 27, 28, 29]
(Fig. 11.10, e-Fig. 11.8). As these lesions display all of the nuclear features of papillary carcinoma, the presence of the hyaline stroma may be overlooked and the sample diagnosed as “papillary carcinoma” [30].
filamentous, and cell borders are indistinct. Romanowsky staining highlights the metachromatic hyaline material, perinucleolar clearing, and cytoplasmic bodies. Intranuclear cytoplasmic inclusions, nuclear grooves, and nuclear overlapping are the rule and well visualized on Papanicolaou staining [26, 27, 28, 29]
(Fig. 11.10, e-Fig. 11.8). As these lesions display all of the nuclear features of papillary carcinoma, the presence of the hyaline stroma may be overlooked and the sample diagnosed as “papillary carcinoma” [30].
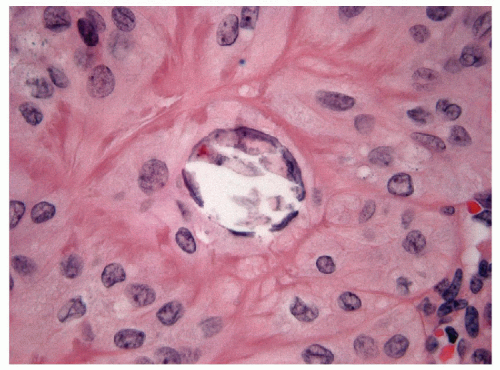 FIGURE 11.7 Hyalinizing trabecular tumor. Psammoma bodies, often numerous, typify hyalinizing trabecular tumors (hematoxylin and eosin stain). |
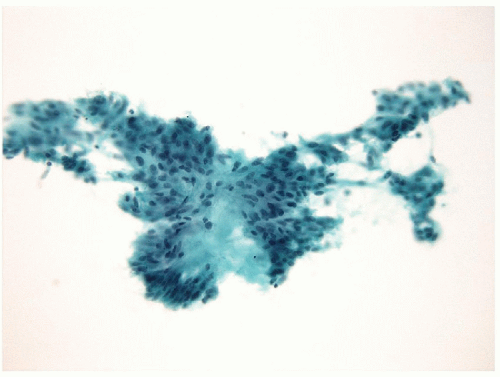 FIGURE 11.8 Fine needle aspiration of hyalinizing trabecular tumor. The tumors characterized by aggregates of hyaline stroma with radial arrangements of spindle epithelial cells (direct smear, Papanicolaou stain). |
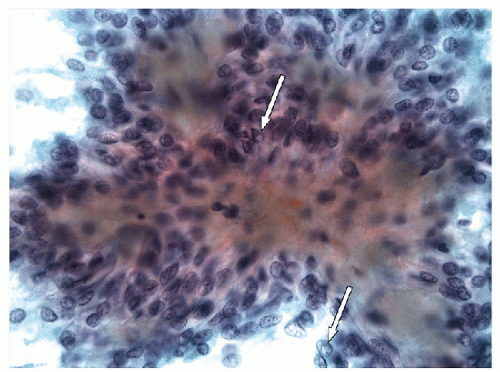 FIGURE 11.9 Hyalinizing trabecular tumor. The epithelial cells of hyalinizing trabecular tumor are elongated, spindle cells, with indistinct cell borders. Even at this power, intranuclear inclusions are readily apparent (white arrows) (direct smear, Papanicolaou stain). |
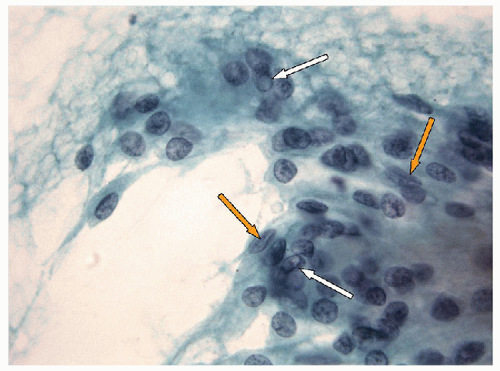 FIGURE 11.10 Hyalinizing trabecular tumor. All of the classical nuclear features of papillary carcinoma are evident and these are typically found in abundance including nuclear grooves (orange arrows) and intranuclear inclusions (white arrows) (direct smear, Papanicolaou stain). |
On histology, the differential diagnosis of these tumors includes paraganglioma or medullary carcinoma [14, 31]. Immunohistochemical stains for neuroendocrine markers easily distinguish HTTs from paraganglioma or medullary carcinoma. HTTs are negative for synaptophysin, chromogranin, and CD56, whereas the neuroendocrine tumors stain for these markers. HTTs stain for thyroglobulin, whereas paraganglioma and medullary carcinoma are negative. CK7 and CK18 have been detected in a high percentage of HTT [32]. Variable expression of CK19 (50% to 100%) (e-Figs. 11.9 and 11.10) has been interpreted by some investigators as proof that these tumors are benign, while others use this as evidence that they should be classified as a variant of papillary carcinoma [32, 33]. Occasional cases are immunoreactive for S100 protein. The identification of anomalous membranous MIB-1 staining in these lesions has been suggested to be of diagnostic assistance [34, 35] (Figs. 11.11 and 11.12). It has been recognized that this staining result is unique to the MIB-1 clone of Ki-67 and is found only if the staining is performed at room temperature. This staining is not present if the procedure is performed at 37°C or other clones for Ki-67 are used [36]. The actual epitope recognized and the biological significance of this unusual staining pattern remains to be clarified.
The marked morphologic similarities between papillary carcinoma and HTT strongly suggest that HHT is simply a variant of papillary carcinoma. Furthermore, application of ret/PTC analysis has identified
rearrangements in these lesions at a rate identical to that found in other papillary carcinomas [21, 22] providing further evidence that this entity is a variant of papillary carcinoma. However, some continue to maintain that these are distinct lesions [32, 34] and the matter remains a subject of debate.
rearrangements in these lesions at a rate identical to that found in other papillary carcinomas [21, 22] providing further evidence that this entity is a variant of papillary carcinoma. However, some continue to maintain that these are distinct lesions [32, 34] and the matter remains a subject of debate.
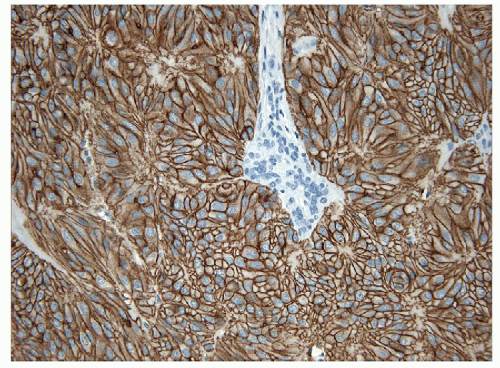 FIGURE 11.11 Hyalinizing trabecular tumor. These lesions show a peculiar membranous staining pattern for MIB-1 (MIB-1 immunoperoxidase stain and hematoxylin). |
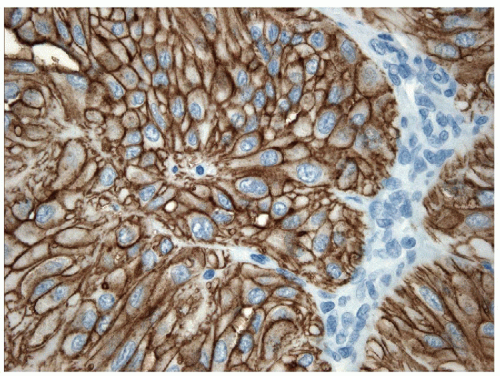 FIGURE 11.12 Hyalinizing trabecular tumor. This anomalous staining for MIB-1 occurs only when the staining is performed at room temperature and with the use of this particular clone and has not been found for other clones of Ki-67. This staining pattern has been suggested as a possible aid to the diagnosis of HTT; however, the actual epitope recognized and the biological significance of this finding have not been established. Note the absence of nuclear staining (MIB-1 immunoperoxidase stain and hematoxylin). |
POORLY DIFFERENTIATED CARCINOMA OF FOLLICULAR EPITHELIAL DERIVATION
Poorly differentiated carcinoma is a tumor of follicular cell origin that occupies a position both morphologically and biologically between well-differentiated papillary or follicular carcinoma and anaplastic thyroid carcinoma [1, 37, 38, 39]. These lesions behave in an aggressive fashion and are often lethal. They do not respond well to treatment with radioactive iodine, since their uptake of this targeted therapy is reduced. This is the lesion that most often is identified as “widely invasive follicular carcinoma” [40]. Vascular invasion and distant metastases are frequent at the time of diagnosis. On complete histological evaluation, there is often a focus of well-differentiated carcinoma (either papillary or follicular) with progression to dedifferentiation; however, some tumors are entirely composed of a poorly differentiated lesion. These tumors may have a central encapsulated nidus, and smaller lesions may not be widely invasive, but usually, the lesion exhibits frank capsular invasion and forms satellite nodules in the surrounding thyroid.
The majority of these lesions have an architectural growth pattern that is characterized by large, well-defined solid nests (Fig. 11.13,
e-Fig. 11.11) that mimics neuroendocrine tumors such as medullary carcinoma; this architecture has given rise to the terminology “insular” carcinoma. Other tumors have trabecular growth patterns. They are largely devoid of follicular architecture or colloid. The tumor cells are usually small and uniform in size (Fig. 11.14, e-Fig. 11.12), and there is mitotic activity with 3 or more mitoses per 10 high-power fields [41, 42] (Fig. 11.15, e-Fig. 11.13). Tumor necrosis is usually identified as single cell necrosis rather than the geographic necrosis that is characteristic of anaplastic carcinoma. In contrast to anaplastic carcinomas, there is little pleomorphism, and no bizarre, giant, or multinucleated cells are found.
e-Fig. 11.11) that mimics neuroendocrine tumors such as medullary carcinoma; this architecture has given rise to the terminology “insular” carcinoma. Other tumors have trabecular growth patterns. They are largely devoid of follicular architecture or colloid. The tumor cells are usually small and uniform in size (Fig. 11.14, e-Fig. 11.12), and there is mitotic activity with 3 or more mitoses per 10 high-power fields [41, 42] (Fig. 11.15, e-Fig. 11.13). Tumor necrosis is usually identified as single cell necrosis rather than the geographic necrosis that is characteristic of anaplastic carcinoma. In contrast to anaplastic carcinomas, there is little pleomorphism, and no bizarre, giant, or multinucleated cells are found.
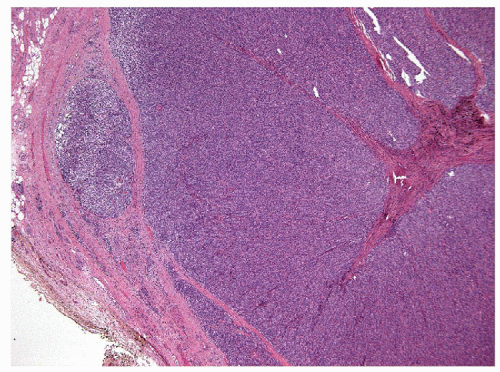 FIGURE 11.13 Poorly differentiated carcinoma. At low magnification this tumor shows a growth pattern composed of solid nests of cells, resembling neuroendocrine tumors and giving origin to the designation of “insular” carcinomas (hematoxylin and eosin stain). |
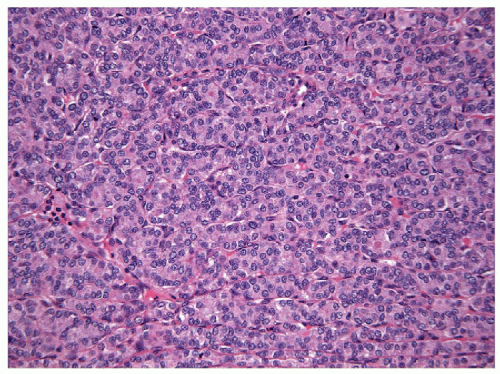 FIGURE 11.14 Poorly differentiated carcinoma. Colloid is typically absent and the constituent cells are comparatively smaller and uniform in their appearance (hematoxylin and eosin stain). |
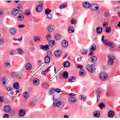
FNA of these lesions yields extremely cellular smears that are devoid of colloid [43, 44, 45, 46]. The tumor is composed of small- to medium-sized epithelial cells seen individually and in small groups and solid clusters (Figs. 11.16, 11.17, 11.18, 11.19, e-Figs. 11.14 and 11.15). Occasionally, a necrotic background is present. The cytoplasm is pale and may contain fine vacuoles. Nuclei are round and monomorphic with fine, hyperchromatic chromatin and variable but often small nucleoli. Although demonstrating a “neoplastic” appearance, there is only mild nuclear atypia. The nuclei tend to be oriented around the periphery of the tumor cell clusters, and there may be basement membrane-like material, transgressing vessels, and endothelial wrapping of cell clusters [47]. In some cases, nuclear inclusions and grooves may be seen and indicate dedifferentiation of a papillary carcinoma.
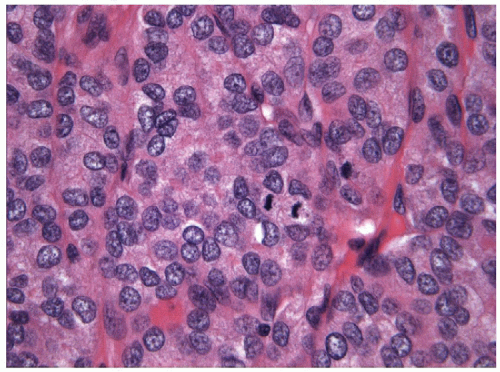 FIGURE 11.15 Poorly differentiated carcinoma. Mitotic activity is easily identified and tumor necrosis in the form of individual cell death is commonplace (hematoxylin and eosin stain). |
The differential diagnosis includes medullary carcinoma of the thyroid and low-grade lymphoma. Sclerosis can mimic amyloid; however, Congo red stains are negative, and immunohistochemical stains for calcitonin, chromogranin, and CEA are negative. Markers of hematologic
differentiation are negative. In contrast, the tumors are positive for keratins, TTF-1, and Pax8 (Fig. 11.20), and staining for thyroglobulin, although often weak and sometimes focal, confirms the follicular cell differentiation of this neoplasm [48].
differentiation are negative. In contrast, the tumors are positive for keratins, TTF-1, and Pax8 (Fig. 11.20), and staining for thyroglobulin, although often weak and sometimes focal, confirms the follicular cell differentiation of this neoplasm [48].
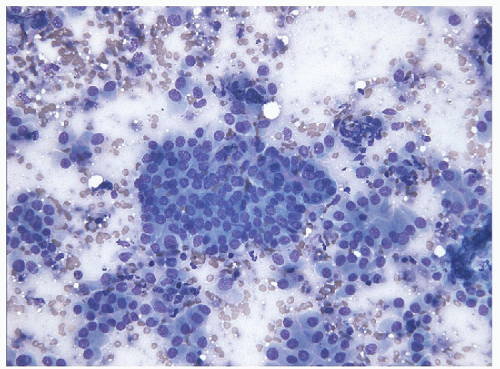 FIGURE 11.16 Poorly differentiated carcinoma. FNAs are typically cellular with the epithelial cells seen individually, in small groups and as solid cell masses (direct smear, air-dried Diff-Quik stain). |
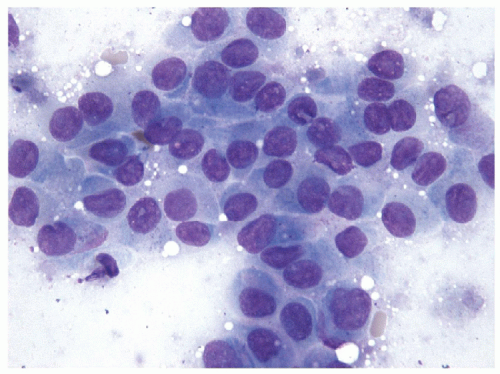 FIGURE 11.17 Poorly differentiated carcinoma. The cells appear relatively monomorphic with cytoplasm containing fine vacuoles, round nuclei, fine, hyperchromatic chromatin and variable but often small nucleoli. Although demonstrating a “neoplastic” appearance, there is only mild nuclear atypia (direct smear, air-dried Diff-Quik stain). |
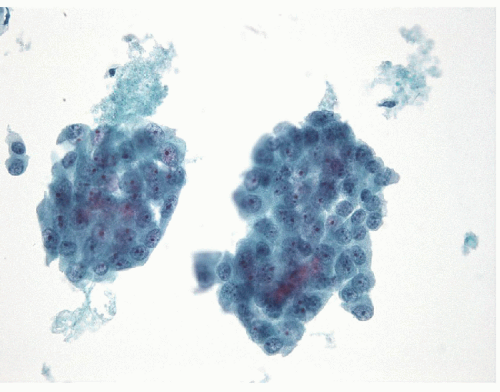 FIGURE 11.18 Poorly differentiated carcinoma. On ThinPrep, the appearance is similar to direct smears with a cellular sample and numerous epithelial cells seen as individual cells and in complex groups. Note the necrotic background (ThinPrep, Papanicolaou stain). |
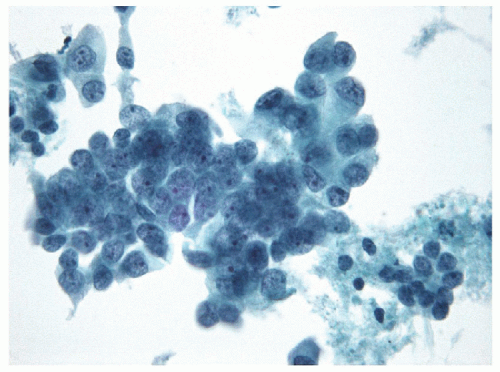 FIGURE 11.19 Poorly differentiated carcinoma. The cellular monomorphism is apparent on ThinPrep samples and helps distinguish these tumors from anaplastic carcinoma of the thyroid (ThinPrep, Papanicolaou stain). |
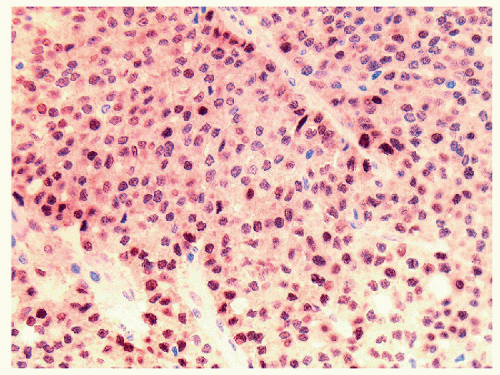 FIGURE 11.20 Poorly differentiated thyroid carcinoma. The solid epithelial cells can mimic many other carcinomas. Nuclear staining for Pax8 (illustrated) and TTF-1, as well as focal cytoplasmic thyroglobulin reactivity, confirms the diagnosis. |
Molecular studies have identified a high incidence of ras mutations in these lesions [49], and up to 25% harbor mutations of the CTNNB1 gene, resulting in nuclear translocation of β-catenin and activation of a number of downstream targets of the WNT signaling pathway [50]. These mutations are not found in well-differentiated thyroid carcinomas and are more common in anaplastic carcinomas, suggesting that they play a role in progression of these lesions [51]. APC mutations, another cause of nuclear translocation of β-catenin, are not reported in thyroid carcinomas [51]. The loss of membranous staining for β-catenin can be seen by immunohistochemistry, and nuclear translocation is a marker of dedifferentiation. E-cadherin is also reduced or lost [52]. The loss of β-catenin and E-cadherin in these tumors may account for the dehiscence that is characteristic of poorly differentiated carcinoma.
Poorly differentiated carcinomas often express HBME-1 [53, 54, 55, 56], but the diagnosis of malignancy that is supported by this stain is not usually controversial in this entity. The expression of CK7, CK18, and CK19 is reduced in poorly differentiated carcinomas [57].
The molecular basis for dedifferentiation is postulated to be due to multiple genetic events. These tumors may have mutations of BRAF or RAS that are found in differentiated thyroid cancers [51, 58]. They have been reported to exhibit a low prevalence of expression of RET rearrangements [51, 54], a finding that has been used as evidence that RET/PTC rearrangements predict indolent behavior; however, it is known that the promoters of the upstream genes involved in the translocations determine variable levels of expression and dedifferentiation results in down-regulation of expression with loss of RET/PTC gene expression by RT-PCR as well as protein expression by immunohistochemistry. In addition to the mutations and rearrangements that occur in differentiated thyroid cancer, they often also harbor additional events, including mutations in PIK3CA [52, 59, 60], CTNNB1 [61], PTEN [61], the CDKI family of cell cycle regulators and p53 [61], AKT [58], TERT [62], and an STRN-ALK rearrangement [63].
Variants
Aggressive solid tumors that have widely invasive growth and are composed of oncocytic cells represent an oncocytic variant of poorly differentiated carcinoma (Fig. 11.21). In our experience, most aggressive oncocytic or Hürthle cell lesions show insular growth and focal tumor cell necrosis; this may explain the poor outcome of tumors diagnosed as Hürthle cell carcinomas [64]. As with other oncocytic lesions, these tumors likely have mutations of mitochondrial DNA in addition to the known mutations of poorly differentiated thyroid carcinomas [65]. The role of PTEN p53 mutations in these oncocytic tumors remains to be determined [66].
Clear cell carcinoma is rare in the thyroid but often has solid growth and, when primary, often represents a clear cell variant of poorly differentiated carcinoma with accumulation of glycogen, lipid, or even mucin [20] (Fig. 11.22, e-Fig. 11.16). The term “clear cell tumor” should be restricted to lesions in which more than 75% of the tumor cells show
this change. Clear cell change may occur focally within oncocytic lesions. The finding of a clear cell tumor raises an important differential diagnoses, since a common differential diagnosis is metastasis, particularly from renal or adrenal tumors [20], or biopsy of adjacent parathyroid tissue. Parathyroid nodules can be adjacent to or within the thyroid and can mimic a primary thyroid lesion [67]. A novel approach using Fourier transform infrared spectroscopic (FTIR) interrogation of touch imprint cytology provides detailed spectra, which can be used to distinguish various tissue pathologies. FTIR spectral profiles of thyroid tissues differ visually when compared with parathyroid tissue [68]. In more routine studies, proof of follicular cell derivation is obtained from thyroglobulin, TTF-1, and Pax8 staining, while other tissues have distinct immunoprofiles.
this change. Clear cell change may occur focally within oncocytic lesions. The finding of a clear cell tumor raises an important differential diagnoses, since a common differential diagnosis is metastasis, particularly from renal or adrenal tumors [20], or biopsy of adjacent parathyroid tissue. Parathyroid nodules can be adjacent to or within the thyroid and can mimic a primary thyroid lesion [67]. A novel approach using Fourier transform infrared spectroscopic (FTIR) interrogation of touch imprint cytology provides detailed spectra, which can be used to distinguish various tissue pathologies. FTIR spectral profiles of thyroid tissues differ visually when compared with parathyroid tissue [68]. In more routine studies, proof of follicular cell derivation is obtained from thyroglobulin, TTF-1, and Pax8 staining, while other tissues have distinct immunoprofiles.
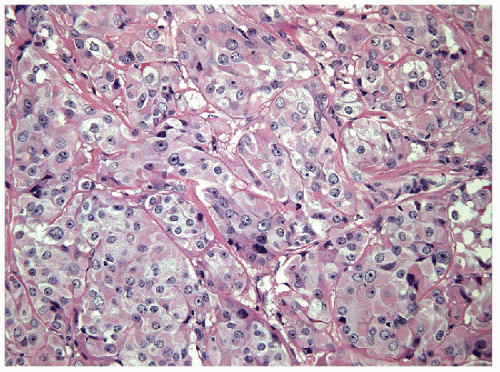 FIGURE 11.21 Oncocytic variant of poorly differentiated carcinoma. Aggressive oncocytic neoplasms that have been classified as “Hürthle cell carcinoma” show insular growth and focal tumor cell necrosis (hematoxylin and eosin stain). |
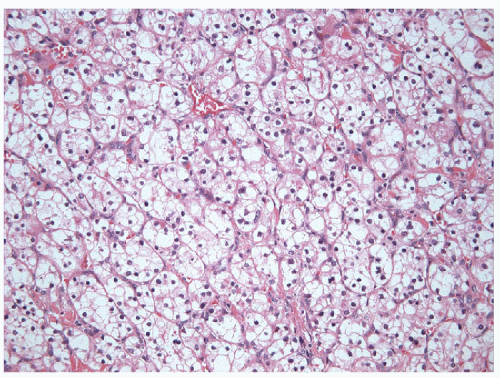 FIGURE 11.22 Clear cell poorly differentiated carcinoma. Focal clear cell change is common in oncocytic neoplasm, but when an excess of 75% of the tumor shows clear cell differentiation, there is the possibility of a clear cell carcinoma of thyroid as seen in this illustration. It is imperative to exclude metastatic clear cell carcinoma (adrenal, renal) and the remote possibility of a clear cell parathyroid lesion (hematoxylin and eosin stain). |
MEDULLARY CARCINOMA
Medullary thyroid carcinoma is a tumor of parafollicular C cells of the thyroid that synthesize and secrete calcitonin. C cells were initially described by Karl Hürthle [69] whose name has mistakenly been associated with oncocytes. The function of C cells only became apparent with the discovery of the hormone calcitonin and the description of medullary thyroid carcinoma [70].
Medullary carcinoma comprises approximately 5% of all thyroid carcinomas [20] but is responsible for 13.4% of the total deaths [71]. This lesion is usually readily recognized because of its unusual cytologic and histologic features, but it is also frequently misdiagnosed [1, 72]. Sometimes special investigation is required to distinguish it from follicular lesions or other tumors, including lymphomas and/or anaplastic carcinomas. The importance of distinguishing this tumor from follicular lesions is twofold. The first is for diagnostic classification and management considerations in the individual patient. These tumors do not preferentially take up iodine, and therapy with radioactive iodine is not indicated; in contrast, expression of somatostatin receptors by some of these tumors [73] makes somatostatinlabeled imaging a feasible diagnostic tool to localize the primary lesion and to identify metastatic deposits [74, 75], and somatostatin analogues as well as somatostatin-based peptide receptor radiotherapy (PRRT) may have applications in the management of disseminated disease [76, 77, 78]. The other aspect of management involves the implications for both the patient and members of his/her family, since many of these tumors are hereditary [79, 80].
The inherited forms of medullary carcinoma are of two major types [80]: multiple endocrine neoplasia (MEN) type IIA in which MTC may be associated with pheochromocytomas or may occur alone (also known as familial medullary thyroid carcinoma, FMTC) and MEN IIB in which the thyroid and adrenal proliferative disorders are associated with mucosal ganglioneuromas and a Marfanoid habitus. Other variants of MEN2A include associations with cutaneous lichen amyloidosis (CLA) and with Hirschsprung’s disease [80]. The inheritance of these syndromes was mapped to the pericentromeric region of chromosome 10 by linkage analysis [81, 82, 83]. Subsequently, mutations in the RET proto-oncogene in
exon 10 (codons 609, 611, 618, and 620), exon 11 (codons 630 and 634), exons 8, 13, 14, 15, and 16 have been identified in patients with MEN IIA and at codon 918 in exon 16 or rarely at codon A883F in exon 15 in MEN IIB [80, 84, 85]; these mutations have provided a more accurate marker of germ-line mutation and predisposition to this disease [71, 86, 87]. Current recommendations suggest that family members of known kindreds have genetic screening early in life and affected members should undergo prophylactic thyroidectomy in childhood [71, 80]. The ages recommended vary depending on the mutation and family history; the goal is to prevent the earliest possible onset of medullary thyroid carcinoma in these familial forms of the disease, since metastatic tumor has been found in patients as young as 6 years of age. Children affected with MEN IIB undergo surgery even earlier than those with MEN IIA and FMTC [88, 89].
exon 10 (codons 609, 611, 618, and 620), exon 11 (codons 630 and 634), exons 8, 13, 14, 15, and 16 have been identified in patients with MEN IIA and at codon 918 in exon 16 or rarely at codon A883F in exon 15 in MEN IIB [80, 84, 85]; these mutations have provided a more accurate marker of germ-line mutation and predisposition to this disease [71, 86, 87]. Current recommendations suggest that family members of known kindreds have genetic screening early in life and affected members should undergo prophylactic thyroidectomy in childhood [71, 80]. The ages recommended vary depending on the mutation and family history; the goal is to prevent the earliest possible onset of medullary thyroid carcinoma in these familial forms of the disease, since metastatic tumor has been found in patients as young as 6 years of age. Children affected with MEN IIB undergo surgery even earlier than those with MEN IIA and FMTC [88, 89].
Familial forms of medullary thyroid carcinoma usually result in multicentric disease as well as multicentric C-cell hyperplasia [90, 91]. C cells are usually limited to the central portion of the junction between the upper and middle thirds of the lateral lobes where they are generally distributed singly rather than in clusters. Increased numbers of C cells (greater than 7 cells per cluster), complete follicles surrounded by C cells, and distribution of cells beyond this geographic location are indicative of C-cell hyperplasia (Fig. 11.23, e-Figs. 11.17 and 11.18). The assessment of C-cell hyperplasia should only be performed on the lobe opposite a large tumor since infiltrating tumor can mimic this entity. The presence of C-cell hyperplasia
usually indicates an inherited disorder rather than a sporadic lesion; however, C-cell hyperplasia can also be associated with chronic hypercalcemia, thyroid follicular nodular disease, and thyroiditis [92, 93, 94, 95]. Patients with PTEN hamartoma tumor syndrome (PHTS), characterized by a predominance of nonthyroidal tumors, have a predisposition to develop multiple adenomatous follicular nodules but have also been reported to have a diffuse form of C-cell hyperplasia [96], but this has not been associated with medullary carcinoma. Drugs, specifically antidiabetic incretins (glucagon-like peptide-1 analogs including exenatide, liraglutide, and taspoglutide), cause C-cell hyperplasia and medullary thyroid carcinoma in rodents [97, 98] where GLP-1 receptors are expressed by C cells; although human medullary thyroid carcinomas may express GIP and GLP-1, normal human C cells do not express their receptors[99]; nevertheless, it has been suggested that patients be monitored with calcitonin measurements during GLP-1 analog administration [99].
usually indicates an inherited disorder rather than a sporadic lesion; however, C-cell hyperplasia can also be associated with chronic hypercalcemia, thyroid follicular nodular disease, and thyroiditis [92, 93, 94, 95]. Patients with PTEN hamartoma tumor syndrome (PHTS), characterized by a predominance of nonthyroidal tumors, have a predisposition to develop multiple adenomatous follicular nodules but have also been reported to have a diffuse form of C-cell hyperplasia [96], but this has not been associated with medullary carcinoma. Drugs, specifically antidiabetic incretins (glucagon-like peptide-1 analogs including exenatide, liraglutide, and taspoglutide), cause C-cell hyperplasia and medullary thyroid carcinoma in rodents [97, 98] where GLP-1 receptors are expressed by C cells; although human medullary thyroid carcinomas may express GIP and GLP-1, normal human C cells do not express their receptors[99]; nevertheless, it has been suggested that patients be monitored with calcitonin measurements during GLP-1 analog administration [99].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


