Soft tissue sarcomas
Robert G. Maki, MD, PhD, FACP  Chandrajit P. Raut, MD, MSc, FACS
Chandrajit P. Raut, MD, MSc, FACS  Brian O’Sullivan, MD, FRCPI, FRCPC
Brian O’Sullivan, MD, FRCPI, FRCPC
Overview
The management of soft tissue sarcomas is driven by the anatomic site and histology of the primary and is increasingly affected by the specific genetics of the sarcoma. In this chapter, we focus on the principles of management of this group of over 50 cancer subtypes to highlight commonalities and differences from anatomical constraints of surgery to specifics of adjuvant radiation to identification of systemic therapeutics that are appropriate for each histology.
We will discuss in this chapter the etiology, presentation, diagnosis, staging, and multidisciplinary management of patients with sarcomas of soft tissue. Surgery remains paramount to achieve cure for the vast majority of sarcomas. Radiation therapy is used for larger tumors in the appropriate clinical context. The evolution of increasingly sophisticated radiation techniques is highlighted in this chapter. As pertains to systemic therapy, this chapter is written at a time in which first-line therapies for soft tissue sarcomas may change, raising anew some questions of adjuvant therapy that remain incompletely answered. Where appropriate, we attempt to link specific histologies or molecular changes to therapeutic suggestions, with the understanding and hope that novel agents will supplant the medications that are available but that have not materially affected outcomes for few diagnoses other than GIST in the past several years.
Sarcomas of nonosseous tissues, known traditionally as soft tissue sarcomas (STS), comprise a group of rare malignancies that exhibit tremendous diversity of anatomic site, specific genetic alterations, and histopathologic characteristics. These tumors share a common embryologic origin, arising primarily from mesodermal tissues. The notable exceptions are sarcomas of the neural tissues [such as malignant peripheral nerve sheath tumors (MPNST)] and possibly the Ewing sarcoma/primitive neuroectodermal tumor (PNET) family of tumors, which are believed to arise from ectoderm and angiosarcomas, which are derived from endoderm. Despite the fact that the somatic nonosseous tissues account for as much as 75% of total body weight, primary neoplasms of these connective tissues are comparatively rare, accounting for <1% of adult malignancies and 15% of pediatric malignancies. About 12,000 people receive a diagnosis of STS in the United States each year, with approximately 5000 deaths annually.1 An understanding of these cancers is important because patients’ outcomes will be compromised if initial management is not thoughtful. Furthermore, biologic insights about sarcomas are providing new strategies for the detection, treatment, and prevention of more common malignancies.
This chapter reviews current concepts in the diagnosis, staging, and multidisciplinary management of patients with sarcomas of nonosseous tissues. The evolving contributions of molecular biology and basic scientific principles underlying the varied differentiation and clinical behavior of these tumors will also be reviewed. Although histopathologic aspects of sarcomas are increasingly important in categorizing these tumors, the anatomic site of primary disease remains an important variable on which treatment and outcome may depend. Extremity sarcomas account for approximately 50% of all sarcomas and are the primary focus of the therapy sections of this chapter. Special topics such as retroperitoneal sarcomas (RPS), gastrointestinal stromal tumors (GISTs), and dermatofibrosarcoma protuberans (DFSPs) are addressed separately later in this chapter. Sarcomas at other anatomic sites are not discussed because of their rarity. Throughout the chapter, the emphasis is on identifying what is known from definitive data and what requires additional research.
Etiology
Most sarcomas are believed to arise spontaneously, as may be increasingly understood for many cancers in general.2 The conceptual frameworks that address the neoplastic transformation of mesenchymal stem cells are in rapid evolution owing to new insights from the molecular analysis of sarcomatous and normal tissues from STS patients and family members. Genetics and environmental factors appear to play a role in the neoplastic transformation of soft tissues into sarcomas.3
It has been recognized for more than 30 years that sarcomas can arise in persons with certain genetic predispositions to cancer development. One of the earliest observations of familial cancer development (i.e., genetically transmitted predisposition to malignancy) was the development of sarcoma and other tumor types (such as breast cancer) in certain families.4 This autosomal dominant genetic predisposition has now become known as the Li–Fraumeni syndrome, and it has been characterized at the molecular level as a germline mutation of the TP53 gene, which presumably acts in this context as a faulty tumor suppressor.5, 6
Other genetic disorders are also associated with an increased risk of developing specific sarcomas. The best-studied example of this is the predilection of patients with neurofibromatoses to develop (MPNSTs, also referred to as neurofibrosarcomas or malignant schwannomas).7, 8 Type 1 neurofibromatosis (von Recklinghausen disease) is an autosomal dominant disease that can disrupt the function of the NF1 gene, located on chromosome 17q11.2. The endogenous function of the NF1 gene product, neurofibromin, remains incompletely understood, but it appears to act as a tumor suppressor via stimulation of guanosine triphosphatase activity. Common mutations in NF1 include truncations, with loss of function leading to uncontrolled signaling through ras pathways, which impact on therapeutic options.9, 10 NF1 loss appears to be a fundamental process that facilitates the development of MPNSTs over time in patients with neurofibromatosis. Patients with type 1 neurofibromatosis have up to a 10% cumulative lifetime risk of developing sarcoma (usually MPNST); it is unclear why the risk is not greater, given the protean effects of ras activation; other factors such as inactivation of epigenetic regulators in the PRC2 complex may contribute to the neoplastic phenotype.11–13
Survivors of childhood retinoblastoma have also been noted to have an increased risk of sarcoma development later in life.14, 15 These data provide another model of a dysfunctional or deleted tumor suppressor genetic element (in this case, the product of the Rb gene on chromosome 13q14). The risk of STS in retinoblastoma patients and their families is accompanied by the risk of developing several other types of neoplasms, including osteosarcomas, breast cancer, and lung cancer. No reasons have been convincingly posited for the development of one type of malignancy over another in patients with Rb mutations, and this remains an important question to be addressed by future research on mechanisms of neoplastic transformation.
Gardner syndrome represents an important genetic connection between dysfunctional regulation of epithelial and mesenchymal cells. Gardner syndrome represents a subset of familial adenomatous polyposis disorders of the bowel (usually the colon); patients with the syndrome also have extracolonic abnormalities such as epidermoid cysts and osteomas. The molecular lesion has been identified as a defect within the APC (adenomatous polyposis coli) gene on chromosome 5q21. Patients with Gardner syndrome are at much increased risk of developing mesenteric and intraperitoneal desmoid tumors.16, 17 Desmoid tumors are mesenchymal cells proliferating in a pattern of aggressive fibromatosis, characterized by bland cells that—although histologically benign—act in a malignant manner with uncontrolled proliferation and infiltration of vital structures; spontaneous desmoid tumors more commonly demonstrate the β-catenin gene, CTNNB1, which is in the same signaling pathway as APC.18, 19 It remains poorly understood why some patients with Gardner syndrome develop desmoid tumors whereas others do not, and the lifetime risk of developing desmoid tumors has been estimated at approximately 10–20%, representing a nearly 1000 times greater risk than that of the general population.
Certain environmental exposures have also been associated with the development of sarcomas. One of the most important is ionizing radiation. Radiation-associated sarcoma is most often a late effect of radiotherapy (RT) given to treat another condition (often a prior malignancy). Sarcomas have been noted as a late effect of RT for breast cancer, Hodgkin lymphoma, non-Hodgkin lymphomas, and other tumor types.20 The radiation dose appears to be correlated with the later development of sarcoma, with a very low risk in patients who received <10 Gy. The molecular mechanisms may be complex, as it has been noted clinically that sarcomas appear at the margins of prior RT fields. This suggests that the mutagenic effect may be maximal at the edges of prior RT where scatter radiation leads to a dose sufficient to induce mutations but insufficient to kill the mutated cells. Traditionally, radiation-associated sarcomas were thought to arise with a median of ∼9 years following RT, although RT-associated sarcomas are observed earlier in some patients. MPNSTs, angiosarcomas, osteosarcomas, and undifferentiated pleomorphic sarcomas (UPS) comprise the majority of radiation-associated sarcomas. Clinical outcomes are worse in patients with radiation-associated sarcomas compared to histology-matched controls. Radiation-associated sarcomas should be approached as new primary disease and treated appropriately to optimize the patient’s outcomes.
Certain chemical exposures have also been weakly linked to sarcomagenesis, although chemical-induced development of sarcomas in animal models is one of the more reliable models of studying neoplastic transformation in the laboratory. Hepatic angiosarcomas are associated with exposure to several classes of chemicals, such as polyvinyl chloride and arsenic compounds.21 The relationship between exposure and development of sarcoma is more tenuous for other compounds, including dioxins (such as Agent Orange and other phenoxyacetic acid-based herbicides) and chlorophenols used in wood preservatives.22
Chronic irritation or inflammation of tissues is a controversial potential cause of sarcomas. Certainly, there is an increased sarcoma risk in the lymphedematous arms of women who have undergone radical mastectomy (the Stewart–Treves syndrome), often with the additional complicating variable of prior RT.23, 24 Limited data, typically case reports only, suggest that other sources of chronic tissue irritation and inflammation might be associated with sarcomagenesis.25 Although a history of trauma is not infrequently elicited from patients with STSs, the impact of such trauma on sarcoma development is dubious.
Severe and chronic immunosuppression following solid organ transplantation represents yet another risk factor for the development of sarcomas. Sarcomas represent a disproportionate percentage of tumors (10%) in patients following solid organ transplantation, with Kaposi sarcoma comprising the majority of these.26, 27
Screening
Given the rarity of sarcomas in the general population, no general screening is indicated beyond routine health care surveillance. However, it is important for physicians to be aware of the predisposing genetic tendencies and environmental exposures that might increase patients’ risk of sarcoma development. A complete family history should reveal clues about genetic predispositions, including a family history of polyposis, neurofibromatosis, retinoblastoma, any cancer at a young age in first-degree relatives, or sarcomas. Genetic counseling is appropriate to discuss issues relating to these predispositions, in particular given the finding of Li–Fraumeni-like families that lack canonical TP53 mutations.28 In patients at increased risk of sarcoma, a more detailed clinical evaluation might be required at a lower threshold of intervention than one might use in general practice. Rapidly growing masses, especially symptomatic ones, in patients with neurofibromatosis should be considered for surgical removal to rule out the potential of sarcomatous transformation of a neurofibroma. Similarly, any superficial or deep abnormalities of skin or soft tissues in patients with a history of prior RT should be evaluated very thoroughly.
Clinical presentation, classification, and diagnosis
Sites of origin
Sarcomas of nonosseous tissues have been noted to arise at virtually all anatomic sites. The anatomic sites and site-specific histologic subtypes of more than 5113 sarcomas treated at a single referral institution are outlined in Figure 1. Approximately one-third to one-half of all sarcomas of nonosseous tissues occur in the lower extremities, where the most common histopathologic subtypes have traditionally been noted to include liposarcomas and the entity “UPS”, formerly termed malignant fibrous histiocytoma (MFH). With improved pathologic tools to categorize sarcomas (e.g., immunohistochemistry, DNA, and RNA analyses), it is increasingly recognized that UPS may have some features in common with poorly differentiated liposarcomas or leiomyosarcomas, as well as other histologic subtypes.29 RPSs comprise 15–20% of all STSs, with well-differentiated/dedifferentiated liposarcoma and leiomyosarcoma being the predominant histologic subtypes. Visceral sarcomas make up an additional 24%, and the head and neck sarcomas approximately 4% of sarcomas.
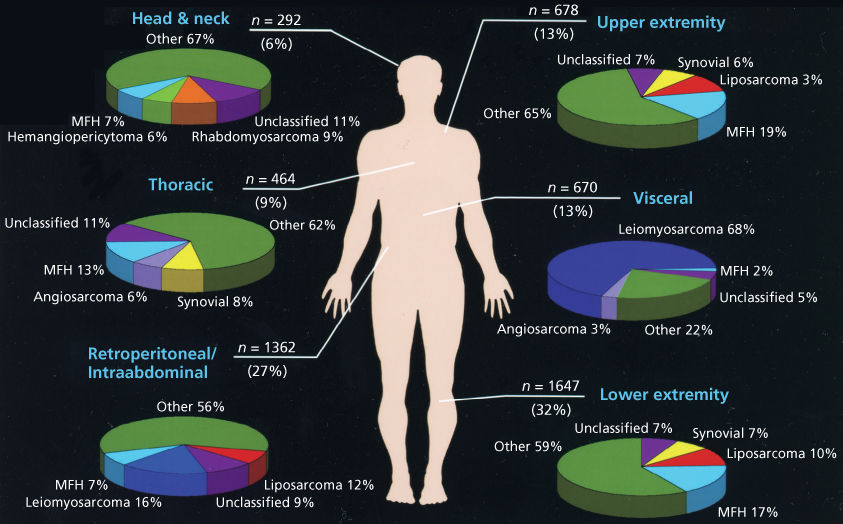
Figure 1 Anatomic distribution and site-specific histologic subtypes of 5113 consecutive STSs seen at the University of Texas MD Anderson Cancer Center Sarcoma Center.
Source: Data from MDACC Sarcoma Database, June 1996 to June 2005.
Clinical presentation
The majority of patients with nonosseous sarcomas present with a painless mass, although pain is noted at presentation in up to one-third of cases.30 Delay in diagnosis of sarcomas is common, with the most common incorrect diagnosis for extremity and trunk lesions being hematoma or “lipoma.” Late diagnosis of RPS is extremely common, as tumors in this area can grow to massive size before causing any symptoms (such as abdominal distention or psoas irritation with back or groin discomfort) or functional compromise such as hydronephrosis from ureteric obstruction.
Physical examination should include an assessment of the size and mobility of the mass. Its relationship to the fascia (superficial vs deep) and nearby neurovascular and bony structures should be noted. A site-specific neurovascular examination and assessment of regional lymph nodes should also be performed. Sarcomas rarely metastasize to lymph nodes, with those that do being limited to a few specific histopathologic subtypes. Presence of true nodal metastases should prompt the clinician to investigate whether the diagnosis of sarcoma is accurate.
Histopathologic classification
Methods of classification
In broad terms, sarcomas can be classified as neoplasms arising in bone versus those arising from the nonosseous or periosseous soft tissues. Sarcomas of nonosseous tissues can be further grouped into those that arise from the viscera (e.g., gastrointestinal or gynecologic organs) and those that originate in nonvisceral soft tissues such as muscle, tendon, adipose tissue, pleura, synovium, and other connective tissues.
The most universally applied classification scheme for STS is based on histogenesis, as outlined in the recently updated WHO (World Health Organization) sarcoma classification system.31 This classification system is reproducible between pathologists for the better differentiated tumors. However, as the degree of histologic differentiation declines, the determination of cellular origin becomes increasingly difficult. For example, pathologists may vary in their criteria to consider a tumor a UPS versus a poorly differentiated leiomyosarcoma; the use of specific DNA tests and ready availability of an increasing battery of immunohistochemical markers have improved consistency in diagnosis. Nonetheless, the lack of familiarity with sarcomas in general leads to misdiagnosis in up to 20% of outside cases reviewed at reference centers.
Difficulties in establishing the specific cellular origin of STS have occasionally been viewed as having limited clinical importance because clinical investigators have not had sufficient data to tie the histologic subtype directly to biologic behavior or to specific therapeutic interventions. Important exceptions to this generalization include epithelioid sarcoma, clear cell sarcoma, angiosarcoma, and embryonal rhabdomyosarcoma, all of which have a greater risk of regional lymph node metastasis.32, 33 In a single-institution study, the overall rate of nodal metastasis at the time of presentation was only 2.7%; however, the rate was much higher for specific histologic subtypes: angiosarcoma (13%), embryonal rhabdomyosarcoma (14%), and epithelioid sarcoma (17%).32 Thus, treatment strategies may differ for these. For the remaining histologic subtypes, biologic behavior appears to be determined more by histologic grade than by histologic subtype. However, as the fundamental biologic and molecular understanding of the mechanisms of malignant transformation in sarcomas increases, in-depth categorization may well prove to have important clinical ramifications. The tools required to categorize or subclassify sarcomas at the molecular level are now increasingly available for many sarcoma subtypes, including GIST, synovial sarcoma, liposarcoma, Ewing sarcoma/PNET, and rhabdomyosarcomas (Table 1). Future clinical trials will need to take histologic and molecular characteristics into account in a more sophisticated manner than in the past three decades of research when markers were not so readily available.
Table 1 Selected cytogenetic aberrations in nonosseous sarcomas
| Histologic subtype | Cytogenetic finding | Genes |
| Myxoid liposarcoma | t(12;16) | FUS-DDIT3 |
| Well-differentiated liposarcoma | Rings and giant markers | Amplified 12q13–15 |
| HMG1C | ||
| CDK4 | ||
| HDM2 | ||
| Lipoma (minimal atypia) | 12q abnormalities | Amplified 12q13–15 |
| Lipoma | 12q14-15 abnormalities | |
| 6p abnormalities | ||
| Synovial sarcoma | t(X;18) | SS18-SSX1, SSX2 or SSX4 |
| Ewing’s family/PNET | t(11;22) and others | EWSR1-FLI1 and others |
| Rhabdomyosarcoma | t(2;13) or t(1;13) | PAX3(or 7)-FOXO1 (alveolar) |
| Clear cell sarcoma | t(12;22) | EWSR1-ATF1 |
| Extraskeletal myxoid chondrosarcomas | t(9;22) | EWSR1-NR4A3 |
| t(9;17) | TAF15-NR4A3 | |
| Dermatofibrosarcoma protuberans | t(17;22) | COL1A1-PDGFB |
| Endometrial stromal sarcoma (low grade) | t(7;17) | JAZF1-SUZ12 |
| Desmoplastic small round-cell tumor | t(11;22) | EWSR1-WT1 |
| Alveolar sarcoma of soft parts | t(X;17) | ASPSCR1-TFE3 |
Abbreviation: PNET, primitive neuroectodermal tumors.
Histologic grade
Biologic aggressiveness can often be predicted based on histologic grade.34 The spectrum of grades varies among specific histologic subtypes (Figure 2). In careful comparative multivariate analyses, histologic grade has been the most important prognostic factor in assessing the risk of distant metastasis and tumor-related death.34, 36 Several grading systems have been proposed, but there is no consensus regarding the specific morphologic criteria that should be employed in the grading of STS.
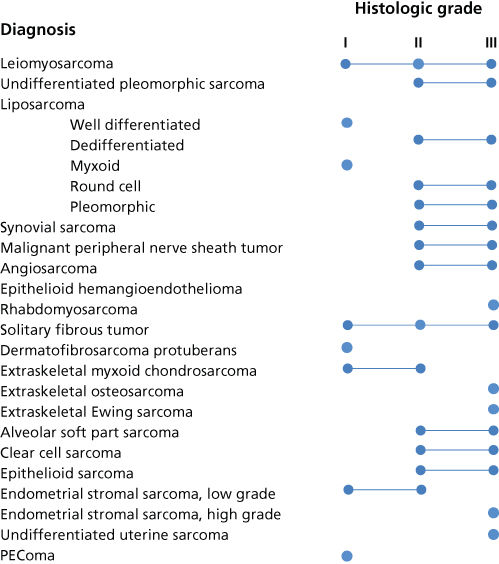
Figure 2 Spectrum of grades observed among histologic subtypes of STS.
Source: From Ref. 35. Adapted from Enzinger FN and Weiss SW, editors. Soft tissue tumors. 5th ed. Mosby-Year Book Inc; 2008.
Two of the most commonly employed grading systems, both first published in 1984, are the US National Cancer Institute (NCI) system developed by Costa and colleagues and the system developed by the Federation Nationale des Centres de Lutte Contre le Cancer (FNCLCC) Sarcoma Group.29, 30, 37 The NCI system is based on the tumor’s histologic subtype, location, and amount of tumor necrosis, but cellularity, nuclear pleomorphism, and mitosis count are also to be considered in certain situations. The FNCLCC system employs a score generated by the evaluation of three parameters: tumor differentiation, mitotic rate, and amount of tumor necrosis. In a retrospective comparison of these two grading systems, in a population of 410 adult patients with nonmetastatic STS, univariate and multivariate analyses suggested that the FNCLCC system has a slightly better ability to predict distant metastasis and tumor-related death.38 Significant discrepancies in assigned grade were observed in one-third of cases. An increased number of grade 3 tumors, reduced number of grade 2 tumors, and better correlation with overall and metastasis-free survival were observed in favor of the FNCLCC system. The FNCLCC system is the best presently available grading system and is employed as part of the AJCC/UICC STS staging system, with the caveat that several new diagnostic categories have been identified since 1984 whose histological grades are undefined by FNCLCC criteria.
In discussing grade, it is important to note well-described characteristics of sarcomas. First, there is often substantial intratumoral heterogeneity within individual sarcomas. Therefore, diagnoses based on very limited amounts of tumor may be inaccurate [e.g., diagnoses based only on fine-needle aspiration (FNA) biopsy specimens]. This is particularly true for such histopathologic subtypes as dedifferentiated liposarcomas, where one area of the tumor might have a relatively low-to-intermediate-grade appearance and another area within the same tumor might have high-grade components more evident. Any discussion of the clinical relevance of grading must take into account this variability inherent in the diagnostic process, which will add to the clinical variability in outcomes among patients with any given grade of sarcomas.
Second, the grade of tumors may evolve over time. This process is best described in the evolution of dedifferentiated liposarcoma arising in conjunction with well-differentiated liposarcoma in the same patient. Additional examples include the round-cell liposarcoma growing from what was previously myxoid liposarcoma and fibrosarcomatous degeneration that will occasionally accompany multiply recurrent DFSPs.
Imaging
Optimal imaging of the primary tumor is dependent on the anatomic site. For soft tissue masses of the extremities, trunk, and occasionally head and neck, magnetic resonance imaging (MRI) generally has been regarded as the imaging modality of choice (Figures 3 and 4) because MRI enhances the contrast between tumor and muscle and between tumor and adjacent blood vessels and provides multiplanar definition of the lesion.39 However, a study by the Radiation Diagnostic Oncology Group that compared MRI and computed tomography (CT) in patients with malignant bone (n = 183) and soft tissue (n = 133) tumors demonstrated no specific advantage of MRI over CT.40 That said, although it may be true that the diagnostic evaluation is equally served by both modalities, surgery and RT planning may require additional information provided by the multiplanar capability of MRI and the ability to perform MRI/CT image fusion.41, 42 For pelvic lesions or evaluation of specific fixed organs, such as the rectum or the liver, the multiplanar capability of MRI may provide superior single-modality imaging (Figure 4), whereas in the retroperitoneum and abdomen, CT usually provides satisfactory anatomic definition of the lesion. Occasionally, MRI with gradient sequence imaging can better delineate the relationship of a tumor to midline vascular structures, particularly the inferior vena cava and aorta (Figure 5). More invasive studies such as angiography or cavography are rarely used in evaluation of STS.
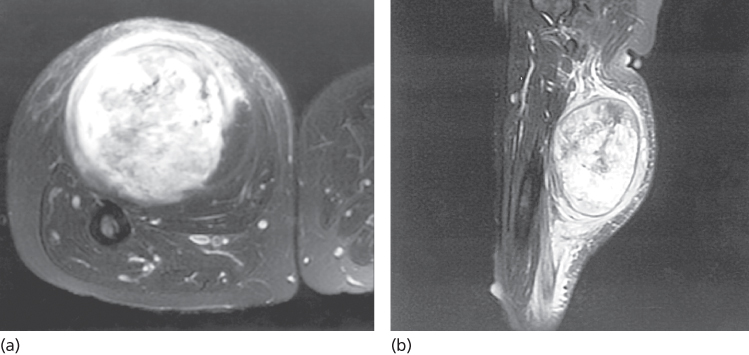
Figure 3 (a) Weighted T2-fat-saturated magnetic resonance image of a TNM T2b high-grade sarcoma in the posterior thigh compartment of a 55-year-old woman. Note the containment by the superficial fascia overlying the posterior thigh muscles, where there is a “strip” of peritumoral edema. Anteriorly, the lesion can be seen to be separate from the femur, but the edge of the tumor is less clearly defined than its superficial component, presumably because of muscle infiltration. (b) Sagittal MRI of the same patient. The main lesion manifests a well-defined border. However, a clear zone of peritumoral edema is evident tracking proximally toward the head of the femur, seen at the top of the figure. Inferiorly, the edema seems to be even more pronounced as evidenced by the triangular signal enhancement pointing inferiorly. Whether the zone of edema harbors microscopic disease is uncertain, and this uncertainty can complicate accurate treatment planning (see text).
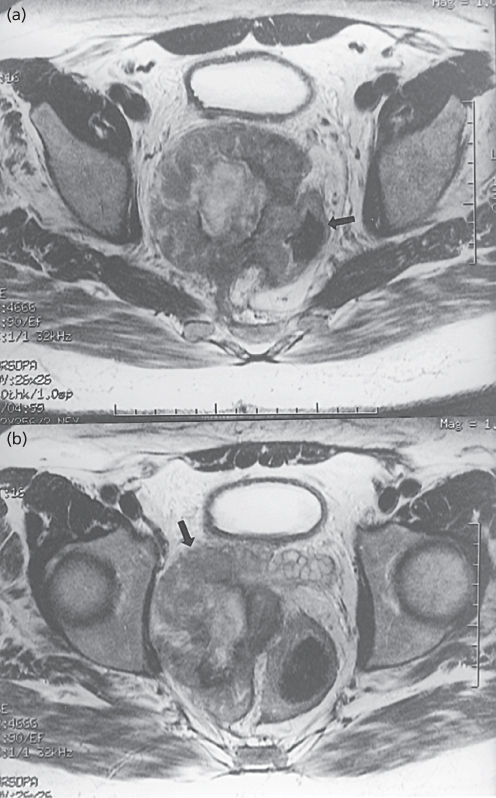
Figure 4 A 57-year-old man with T2 pelvic leiomyosarcoma. (a) Axial T2-weighted fast spin-echo MRI reveals a heterogeneous mass involving the rectum (arrow, air in rectal lumen). (b) Note that the mass abuts right seminal vesicle (arrow).
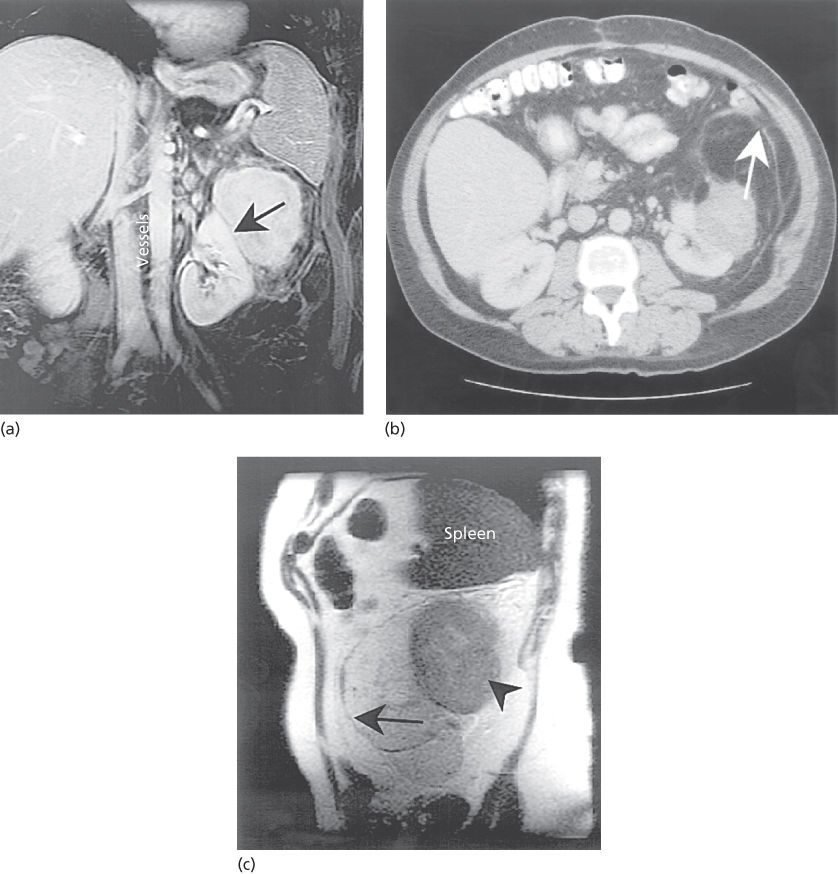
Figure 5 (a) Coronal fat-saturated gadolinium-enhanced MRI showing a solid liposarcoma, 8.3 cm × 6.6 cm, adjacent to and compressing the upper pole of the left kidney. The mass lies below the spleen and is separate from the kidney (line of demarcation, arrow), but is part of a larger fatty tumor. The midline vessels are well visualized. (b) CT image of the same lesion. The mass can be seen adjacent to the kidney, as before. An additional mass of fatty attenuation with gray areas of edema, inflammation, or increased cellularity can be seen bounded by a rim anteriorly (arrow). This mass has the appearance of abnormal fat, which must be considered in treatment planning. Note the displacement of the bowel containing contrast. (c) Sagittal MR image of the same case but without gadolinium. The potential advantage of MR imaging in separating the anterior edge of the retroperitoneal sarcoma (long arrow) from the normal fat anteriorly is seen. The more solid component can also be seen (arrowhead) inferior to the spleen. In addition, these images can be exported digitally to a three-dimensional RT treatment planning workstation or CT simulator workstation where the MR images can be fused to the CT planning slices. This can provide more accurate demonstration of tumor in selected cases for contouring the GTV and clinical target volume than may be possible with CT images alone. This is particularly helpful in situations where CT as well as MRI does not show tumor.
Cost-effective imaging to exclude the possibility of distant metastatic disease is dependent on the size, grade, and anatomic location of the primary tumor. In general, patients with low- and intermediate-grade tumors or high-grade tumors 5 cm or less in diameter require only a chest radiograph for satisfactory staging of the chest. This directly reflects the comparatively low risk of presentation with pulmonary metastases in these patients.43, 44 However, patients with high-grade tumors larger than 5 cm (T2) should undergo more thorough staging of the chest by CT owing to the increased risk of presentation with established metastatic disease in this group.44, 45 Patients with RPS and intra-abdominal visceral sarcomas should undergo imaging of the liver to exclude the possibility of synchronous hepatic metastases; the liver is a more common site of first metastasis from these lesions. CT is usually adequate in these patients to assess the liver, although the increased sensitivity of MRI of the liver may be valuable if any questionable findings are noted on initial CT.
Positron emission tomography (PET) scans may be used selectively to look for extent of disease, particularly when evaluating an ambiguous lesion that could represent a potential metastasis noted on other imaging. However, PET scans are not routinely utilized in staging work-up of STS.
Biopsy
Biopsy of the primary tumor is essential for most patients presenting with soft tissue masses. In general, any soft tissue mass in an adult that is enlarging (even if asymptomatic), is larger than 5 cm, or persists beyond 4–6 weeks should be biopsied. The preferred biopsy approach is generally the least invasive technique required to allow a definitive histologic diagnosis, assessment of grade. In most centers, core-needle biopsy provides sufficient tissue for diagnosis and results in substantial cost savings compared with open surgical biopsy.46, 47 When core-needle biopsy yields insufficient tissue for diagnosis, incisional biopsy is considered to yield optimal amounts of tissue to assess histopathology over a larger area of tumor volume, given the known heterogeneity of sarcomas, as well as to provide sufficient material for detailed molecular and cytogenetic assays. Direct palpation can be used to guide needle biopsy of most superficial lesions, but less accessible sarcomas often require imaging-guided biopsy for safe percutaneous sampling of the most radiographically suspicious area(s) of the mass. Tumor recurrences within the needle track after percutaneous biopsy are exceedingly rare but have been reported, leading some physicians to advocate tattooing the biopsy site for subsequent excision. FNA generally does not provide sufficient material for initial diagnosis, but can be used to confirm recurrence or metastatic disease. Exceptions to this idea exist; endoscopic ultrasound-guided FNA for visceral sarcomas such as GISTs may provide enough tissue for diagnosis while minimizing risk of tumor rupture; in this scenario, it is not feasible to assess mitotic rate. The need for sufficient tissue to conduct more specific molecular testing is a final major rationale for use of core-needle biopsy over FNA. Another major limitation of FNA (compared to core-needle biopsy) is that there is no semblance of preserved tissue architecture to evaluate characteristics such as degree of tissue necrosis.
A practical approach for biopsy and staging of the patient who presents with a primary extremity soft tissue mass is outlined in Figure 6. Small (<5 cm) superficial lesions on an extremity where the morbidity of excisional biopsy is minimal (i.e., remote from joints, tendons, and neurovascular structures that would compromise the surgical margin) are easily biopsied by excisional biopsy with microscopic assessment of surgical margins. For extremity lesions, incisions used for excisional biopsies should be oriented longitudinally along the length of the limb. T2 lesions, T1 lesions located beneath the investing fascia of the extremity, or superficial T1 lesions situated in proximity to joints, tendons, or neurovascular structures are best biopsied by percutaneous core-needle biopsy.
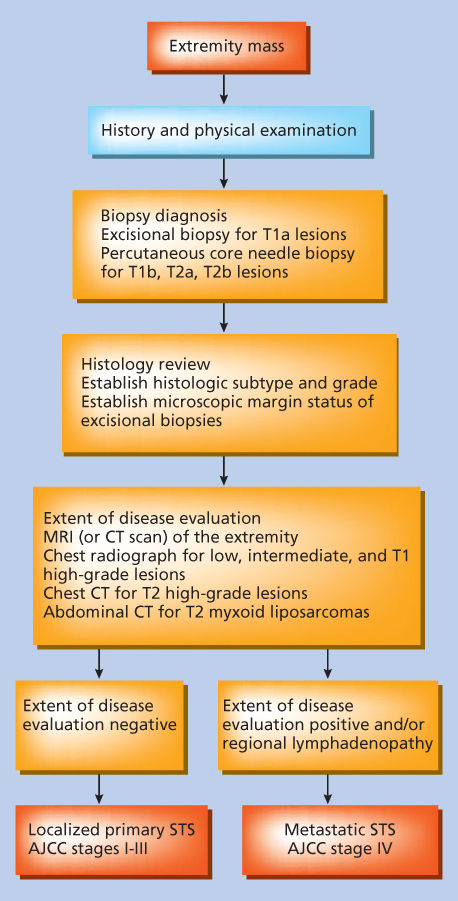
Figure 6 Approach for pretreatment evaluation and staging of the patient presenting with a primary extremity soft tissue mass. AJCC, American Joint Committee on Cancer.
Source: Pisters 1998.48 Reproduced with permission of Springer.
Staging and prognostic factors
Staging
The relative rarity of STS, the anatomic heterogeneity of these lesions, and the presence of more than 50 recognized histologic subtypes of variable grades have made it difficult to establish a functional system that can accurately stage all forms of this disease. The staging system (7th edition) of the American Joint Committee on Cancer (AJCC) and the Union for International Cancer Control is the most widely employed staging system for STS ( Table 2).49 The system is designed to optimally stage extremity tumors but is also applicable to torso, head and neck, and retroperitoneal lesions; a separate staging system is provided for GISTs.
Table 2 American Joint Committee on cancer staging system for STSs, 7th edition
| TX | Primary tumor cannot be assessed | |||||
| T0 | No evidence of primary tumor | |||||
| T1 | Tumor 5 cm or less in greatest dimension | |||||
| T1a | Superficial tumor | |||||
| T1b | Deep tumor | |||||
| T2 | Tumor more than 5 cm in greatest dimension | |||||
| T2a | Superficial tumor | |||||
| T2b | Deep tumor | |||||
| N1 | Regional lymph node metastasis | |||||
| G1 | Well differentiated | |||||
| G2 | Moderately differentiated | |||||
| G3 | Poorly differentiated | |||||
| G4 | Poorly differentiated or undifferentiated (four-tiered systems only) | |||||
| Stage I | T1a, 1b, 2a, 2b | N0 | M0 | G1–2 | G1 | Low |
| Stage II | T1a, 1b, 2a | N0 | M0 | G3–4 | G2–3 | High |
| Stage III | T2b | N0 | M0 | G3–4 | G2–3 | High |
| Stage IV | Any T | N1 | M0 | Any G | Any G | High or low |
| Any T | N0 | M1 | Any G | Any G | High or low | |
Source: Edge et al. 2010.49 Reproduced with permission of Springer.
A major limitation of the present staging system is that it does not take into account the anatomic site of STS. Anatomic site, however, has been recognized as an important determinant of outcome.50, 51 Therefore, although site is not a specific component of any present staging system, outcome data should be reported on a site-specific basis, when feasible. Furthermore, the staging system also fails to include histology, a critical prognostic factor.
Conventional prognostic factors
A thorough understanding of the clinicopathologic factors known to impact outcome is essential in formulating a treatment plan for the patient with STS. Several multivariate analyses of prognostic factors for patients with localized sarcoma have been reported.52–54 However, with few exceptions, most studies have analyzed fewer than 300 patients.
The largest studies established the clinical profile of what is now accepted as the high-risk patient with extremity STS: the patient with a large (≥5 cm), high-grade, deep lesion. In addition, unappreciated prognostic significance includes specific histologic subtypes, for example, MPNST, and the increased risk of adverse outcome associated with a microscopically positive surgical margin or presentation with locally recurrent disease. The type of microscopically positive surgical margins also appears important. Patients with low-grade liposarcomas have a relatively low risk of local recurrence (LR), as do those patients in whom the positive margin is planned before surgery to preserve critical structures and RT can sterilize the small amount of residual disease. However, patients with two categories of positive margin remain at relatively higher risk of LR. These include patients who underwent “unplanned” excision and still have positive margins on re-excision and those with unanticipated positive margins after primary resection.55 An “unplanned excision” is defined as an excisional biopsy or resection carried out without adequate preoperative staging or consideration of the need to remove normal tissue around the tumor.
Unlike for other solid tumors, the adverse prognostic factors for LR of an STS are distinct from those that predict distant metastasis and tumor-related death.52 In other words, patients with a constellation of adverse prognostic factors for LR are not necessarily at increased risk of distant metastasis or tumor-related death. Therefore, staging systems that are designed to stratify patients for risk of distant metastasis and tumor-related death will not necessarily stratify patients for risk of LR.
Kattan and colleagues from the Memorial Sloan-Kettering Cancer Center (MSKCC) have utilized a database of over 2000 prospectively followed adult patients with STS to predict the probability of sarcoma-specific death by 12 years.51 The results have been used to construct and internally validate a nomogram to predict sarcoma-specific death (Figure 6); this and similar nomograms have been validated in a variety of clinical situations, for example, RPS, or for disease-specific contexts, for example, liposarcoma, for individual patients.56–59 These tools may be used for patient counseling, follow-up scheduling, and clinical trial eligibility determination.
Potential molecular prognostic factors
Specific molecular parameters evaluated for prognostic significance in STS have included TP53 mutation, MDM2 amplification, Ki-67 status, altered expression of the RB gene product in high-grade sarcomas, and histologic grade, but not SS18-SSX fusion type, which appears to be an important prognostic factor in patients with synovial sarcoma.60 Complete discussion of the extensive literature on molecular prognostic factors in sarcoma is beyond the scope of this chapter. Readers are referred to more detailed reviews.61, 62
As an example of the difficulty of using even the most commonly recognized markers as prognostic factors, one needs to look no further than Ki-67, an antigen expressed throughout the majority of the cell cycle. Ki-67 is used as a measure of the fraction of cells undergoing division. Preliminary reports of series of heterogeneous sarcomas in adults suggested that Ki-67 nuclear staining correlated with histologic grade, but was not an independent prognostic factor when histologic grade was taken into account.63 Conversely, additional studies in larger numbers of patients indicated that Ki-67 status was an independent prognostic factor for clinical outcomes.64 It is only with the development of consensus guidelines regarding the nature of Ki-67 immunohistochemistry and its interpretation that we can expect to see more careful accurate assessment of this biomarker in sarcoma outcomes.65 It is also with the inconsistencies in the Ki-67 data that one can extrapolate the difficulty of using increasingly available genetic markers for outcome determination. Although specific protein, DNA, and RNA parameters have been identified as having independent prognostic significance, there is presently no consensus on how these prognostic factors should be used in clinical practice.
Treatment of localized primary disease of the extremities
Surgery
General issues
Surgical resection remains the cornerstone of therapy for localized primary STS. The discussion that follows focuses on STSs in the limbs, the most common site of origin, but the principles are equally applicable to sarcomas of other primary anatomic sites.
With the development of limb-sparing techniques in the 1970s and 1980s, there was a marked decline in the rate of amputation as the primary therapy for extremity STS. Today, the widespread application of multimodality treatment strategies means that the vast majority of patients with localized STS of the extremities undergo limb-sparing, usually function-sparing treatment; fewer than 10% of patients presently undergo amputation.66, 67 In selected patients, limb sparing can be approached with surgery alone.
Amputation
Most surgeons consider definite major vascular, bony, or nerve involvement by STS as relative indications for amputation. Complex en bloc bone, vascular, and nerve resections with interposition grafting can be undertaken, but the associated morbidity is high. Therefore, for a few patients with critical involvement of major bony or neurovascular structures, for example, in the foot, amputation remains the only surgical option, but offers the prospect of prompt rehabilitation with excellent local control and survival rates. Other indications for amputation include tumor fungating through the skin or associated with a pathologic fracture with lack of reasonable salvage option.
Combined-modality limb-sparing treatment
Currently, at least 90% of patients with localized extremity sarcomas can undergo limb-sparing procedures. The use of limb-sparing multimodality treatment approaches for extremity sarcoma stems from phase 3 trial from the NCI published in 1982, in which patients with extremity sarcomas amenable to limb-sparing surgery were randomly assigned to receive amputation or limb-sparing surgery with postoperative RT.68 The arms of this trial included postoperative chemotherapy with doxorubicin, cyclophosphamide, and methotrexate. With over 9 years of follow-up, this study established that for patients for whom limb-sparing surgery is an option, limb-sparing surgery combined with postoperative RT and chemotherapy yielded disease-related survival rates comparable to those for amputation while simultaneously preserving a functional extremity.
Satisfactory local resection involves resection of the primary tumor via a longitudinally oriented incision with a margin of normal tissue. Dissection along the tumor pseudocapsule (enucleation) is associated with LR rates in one-third to two-third of patients. In contrast, wide local excision with a margin of normal tissue around the lesion is associated with LR rates in the range of 10–31%, as noted in the control arms (surgery alone) of randomized trials evaluating postoperative RT and in single-institution reports.69
In the modern era, a discussion of limb-preserving approaches must be linked to a discussion of the role of adjuvant therapies, most commonly RT. Several randomized controlled trials have addressed issues surrounding the use of adjuvant therapy and collectively have established important milestones in the evolution of the local management of STS. With a single exception, these trials have focused on extremity lesions and the themes of surgery and adjuvant RT.
Yang et al.70 randomized 91 patients with high-grade extremity lesions following limb-sparing surgery to receive adjuvant chemotherapy alone or concurrent chemotherapy and RT. An additional 50 patients with low-grade tumors were to receive adjuvant RT or no further treatment following limb-sparing surgery. The local control rate for those who received RT was 99% compared with 70% in the no-RT group (p = 0.0001). The results were similar for high- and low-grade tumors (Table 3).
Table 3 Phase 3 trials of adjuvant radiotherapy for localized extremity and trunk sarcoma stratified by grade
| Histologic grade | First author/institution (references) | Treatment group | Radiation dose, Gy | Number of patients | Number of local failure (%) | LRFS (%) | OS (%) |
| High grade | Pisters/MSKCC69 | Surgery + BRT | 42–45 | 56 | 5 (9) | 89 | 27 |
| Surgery | — | 63 | 19 (30) | 66 | 67 | ||
| Yang/NCI70 | Surgery + EBRT | 45 + 18 (boost) | 47 | 0 (0) | 100 | 75 | |
| Surgery | — | 44 | 9 (20) | 78 | 74 | ||
| Low grade | Pisters(/MSKCC69 | Surgery + BRT | 42–45 | 22 | 8 (36) | 73 | 96 |
| Surgery | — | 23 | 6 (26) | 73 | 95 | ||
| Yang/NCI70 | Surgery + EBRT | 45 + 18 (boost) | 26 | 1 (4) | 96 | NR | |
| Surgery | — | 24 | 8 (33) | 63 | NR |
Abbreviations: BRT, brachytherapy; LRFS, local recurrence-free survival; MSKCC, Memorial Sloan-Kettering Cancer Center; NCI, National Cancer Institute; NR, not reported; OS, overall survival; EBRT, external-beam radiotherapy.
Adjuvant RT was also evaluated in a randomized trial of 126 cases treated between 1982 and 1987 (Table 3).69 Brachytherapy (BRT) was administered postoperatively, via an iridium-192 implant that delivered 42–45 Gy over 4–6 days. At 5 years, the local control rate for high-grade tumors was 91% with BRT compared with 70% in surgery-alone controls (p = 0.04). Of note, no improvement in local control with BRT was evident for the low-grade tumors (the local control rate was 74% with surgery alone and 64% with BRT). The full explanation for grade-specific differences in local control with BRT remains unresolved, although one suggestion implicates the relatively long cell cycle of low-grade tumors: low-grade tumor cells may not enter the radiosensitive phases of the cell cycle during the relatively short BRT time. Additional discussion of the pros and cons of BRT compared to external beam radiotherapy (EBRT) is included in the section on “Methods of Radiotherapy Delivery”.
Satisfactory surgical margins to omit radiotherapy
There are no randomized data to define what constitutes a satisfactory gross resection margin for a sarcoma. In general, every effort should be made to achieve a wide margin (2 cm is often an arbitrary choice) around the tumor mass, except in the immediate vicinity of functionally important neurovascular structures, where, in the absence of frank neoplastic involvement, dissection is performed in the immediate perineural or perivascular tissue planes. Technical details of the surgical approach to extremity sarcomas are beyond the scope of this chapter, but are reviewed elsewhere.71 The principle remains that adequate clearance of potential tumor-bearing tissues can be achieved if there is sufficient distance between the surgical margin and the edge of any grossly evident tumor (e.g., at least 2 cm for the closest margin), or where an intact barrier to tumor spread is excised en bloc with the tumor. In such cases, there is little evidence that RT is required even when potential adverse prognostic factors, such as large high-grade tumors are present. The exception in cases of “unplanned” excision where significant contamination of surrounding tissues may have taken place and the precise extent of the tumor is essentially unknown. Depending on the histology, margins of <2 cm are reasonable when an appropriate biological barrier (such as muscle fascia) constitutes that margin. Histologies with infiltrative borders, such as myxofibrosarcoma, may require wider margins or resection. On the other hand, tumors with good prognoses, such as well-differentiated liposarcoma/atypical lipomatous tumor, may be managed by a more limited, marginal resection.
Management of regional lymph nodes
Given the low (2–3%) prevalence of lymph node metastasis in adults with sarcomas, there is no role for routine regional lymph node dissection in most patients.32 However, patients with angiosarcoma, embryonal/alveolar rhabdomyosarcoma, and epithelioid sarcoma have an increased incidence of lymph node metastasis and should be carefully examined for lymphadenopathy. These patients may benefit from the inclusion of lymph node regions electively in adjuvant RT fields.
For patients with STS, lymph node metastasis has been regarded as a particularly adverse finding conferring similar risk to distant metastasis in the TNM (tumor–nodes–metastasis) stage classification. Nevertheless, therapeutic lymph node dissection results in a 34% actuarial survival rate, and thus the rare patients with regional nodal involvement who have no evidence of extranodal disease should undergo therapeutic lymphadenectomy.32 Although formerly classified as being as prognostically as adverse as distant metastasis in the TNM staging classification, isolated lymph node metastasis (as opposed to synchronous distant metastasis), if treated intensively, appears to have a prognosis similar to patients with stage III tumors (i.e., those with high-grade, deep lesions, and lesions larger than 5 cm). The impact of isolated nodal disease was adjusted in the AJCC/UICC staging system by moving N1 disease alone into the stage III group.49 The validity of including N1 disease into stage III was questioned in a follow-up manuscript, in which survival for N1 patients more closely parallels survival with stage IV than node negative AJCC stage III disease.72
Radiotherapy
Rationale for combining radiotherapy with surgery
The use of RT in combination with surgery for STS is supported by two phase 3 clinical trials (Table 3) and is based on two premises: microscopic nests of tumor cells can be destroyed by RT, and less radical surgery can be performed when surgery and RT are combined.69, 70 Although the traditional belief was that STSs were resistant to RT, radiosensitivity assays performed on sarcoma cell lines grown in vitro have confirmed that the radiosensitivity of sarcomas is similar to that of other malignancies; this confirmation supports the first premise.73, 74 The second premise stresses the philosophy of preservation of form (including cosmesis where possible) and function as a goal for many patients with extremity, truncal, breast, and head and neck sarcomas.75–77 Similar principles govern the frequent use of RT for sarcomas at problematic sites such as, for example, RPS, high-risk sarcomas of the head and neck with skull base invasion, or spinal canal invasion by paravertebral lesions. While the efficacy of RT has been confirmed through prospective randomized clinical trials for extremity sarcomas, it has not been confirmed for other sites. Currently, there is an ongoing phase III trial evaluating preoperative RT for primary RPS.
Visceral sarcomas are not ordinarily managed with RT, in part because of the mobile nature of these structures within the pelvic, abdominal, or thoracic compartments. After resection of visceral sarcomas, accurate identification of the field at risk of residual disease is particularly problematic. Contaminated loops of the bowel or mesentery may relocate remotely within the abdominal cavity after surgery, and pleural contamination and mediastinal shift may occur following intrathoracic resections. Fixed tumors in the pelvis or tumors attached to internal truncal walls may occasionally be suited to preoperative or postoperative RT. Typically, however, the vast size of the radiation fields needed to cover entire body cavities, coupled with the limited RT doses that can be safely administered to the organs within the cavities, and the overwhelming risk of distant rather than LR, confines adjuvant RT for the investigational setting.
Essential elements in treatment planning of external beam radiotherapy
Accurate tumor localization is the first essential for RT planning. It primarily uses CT for dosimetric reasons, but MRI can provide complementary information about the tumor extent and can be assimilated in the computer planning workstation through image fusion technology.41, 42 Further essential information is obtained from the pathology and operative reports, and metallic clips placed at the time of surgery may also help define the tumor bed.
It is usually helpful to secure the targeted area to minimize setup variations and eliminate movement during treatment. Simple maneuvers such as comfortable limb positioning or fashioning of customized thermoplastic molds for immobilization will facilitate reliable and consistent treatment setups. RT of superficial tissues, including the scar following definitive resection, with appropriate application of tissue-like bolus material should be considered, but with the recognition that fibrosis, atrophy, and telangiectasias may result. Traditionally, dose uniformity within irregular volumes was optimized using beam segmentation, compensators, or wedge filters. However, this has now largely been replaced by the use of intensity-modulated radiotherapy (IMRT), which addresses issues that include the size, shape, and location of targets and the nature of the normal tissues surrounding them including their contour and width. IMRT is particularly useful in situations where the target volume is adjacent to critical normal tissues as found at the skull base or within the abdomen. Whenever possible, the entire limb circumference, whole joints, or pressure areas (e.g., elbow or heel) should not be treated with what is considered to be a full RT dose, as this may adversely affect limb function and cause distal edema.
It is also prudent to assess baseline function before initiating RT. This is especially important with paired organs, such as eyes or kidneys, if the functional ablation of one organ by RT is expected and is a frequent problem in treating RPS. If right sided or of great size, an RPS may infiltrate the liver capsule or be “hooded” by the liver, making RT access to an appropriate tissue volume surrounding the tumor extremely difficult. This area may be particularly appropriate for IMRT approaches because of the exquisite conformality that is possible with this approach and permits the liver and the other normal tissue to be excluded from the irradiated volume.78, 79 Fortunately, although the tolerance of the entire liver to radiation is low, part of the liver may be safely treated to much higher doses. In these instances, if a subsequent liver resection is needed because of tumor infiltration or adherence to the capsule, detailed consultation between the surgical and radiation oncology teams is needed to ensure that an adequate volume of nonirradiated liver remains in situ.
Dose fractionation issues
Total radiation doses administered postoperatively for sarcoma depend on the tumor grade and involvement of the surgical margin.70, 80, 81 Typical total doses are 60 Gy for low grade and 66 Gy for high-grade tumors, respectively. When RT is given preoperatively, the total dose used in most institutions is approximately 50 Gy in daily fractions administered over 5 weeks.81, 82 However, data regarding radiation dose response are very limited and based on underpowered retrospective studies. On the basis of the current data, higher doses of RT are probably indicated in the postoperative setting (compared with preoperative RT), but the search for an alternative lower dose postoperative schedule seems desirable. These are discussed below in relation to the volumes to be used and the consequences of using different doses in terms of potential morbidity.
The fraction size used in conventional fractionation schemes varies (usually 1.8 or 2.1 Gy).81, 83 Absence of late effects can be expected with smaller fraction size; this tissue is particularly important when critical structures are irradiated. Several altered fractionation schemes have been described including hyperfractionated, hypofractionated, and accelerated schedules.84–87 Most recently, preoperative hypofractionated RT for extremity and trunk wall STS was recently evaluated in a series of 272 patients. RT was delivered preoperatively for 5 consecutive days in 5 Gy per fraction. The LR rate was higher (19%) with the hypofractionated schedule compared to many contemporary series.88 Longer follow-up of this novel strategy is warranted. Neither hyper- nor hypofractionation regimens are likely to replace conventional daily fractionation in the near future, for a combination of reasons that include the small nonrandomized nature of studies, resources needed for some protocols, concerns about efficacy and toxicity, and the fact that modern targeting techniques with IMRT may offset some of the potential benefits that underpin the choice of altered fractionation protocols.
Radiation dose and target volumes
Guidelines have recently been published on how to address the technical design of the radiation volumes and should be discussed for additional detail regarding this topic.89
Many STS respect barriers to tumor spread in the axial plane of the extremity, such as bones, interosseous membranes, or major fascial planes. Consequently, extremity STS tend to spread longitudinally within the specific muscle groups of the extremity. Therefore, the margins of the RT volume must be wide in the cephalocaudal direction. In the cross-section, there may be much greater security in defining nontarget structures, especially those delimited by an intact barrier to tumor spread. Bone, interosseous membranes, and fascial planes are considered barriers to tumor spread in the axial direction, and, therefore, descriptions of radiation margins employed are principally in the cephalocaudal direction. For nonextremity lesions, the preferred direction of spread is also along the direction of the involved musculature, but care must be taken to ensure that the fascial planes are appropriately recognized and encompassed in the radiation target volume.
Earlier, this chapter summarized principles concerning anatomic planes and the preferential pathways for sarcomas to spread within tissues. This information facilitates the design of target areas for RT. The basic elements in RT planning are to first define a gross tumor volume (GTV) and then place a margin around it to encompass tissues at risk of harboring microscopic residual disease [clinical target volume (CTV)] (Figures 7a–c and 8).90 Generally, RT is phased so that an initial volume (phase 1) around the risk zone is treated to doses that are capable of sterilizing microscopic amounts of tumor cells (e.g., 45–50.4 Gy in 1.8 or 2.0 Gy fractions). When delivering RT postoperatively, it is customary to have at least one field reduction to permit an augmented dose to a smaller volume surrounding the highest risk zone (phase 2). This dose is usually 15–16 Gy but can be higher if there is gross residual disease. For the phase 1 volume, the surgical bed is expanded with a 1.5-cm radial margin and a 4-cm craniocaudal margin to encompass microscopic disease in the surrounding tissues; the boost is applied to the original sarcoma localization with a 1.5-cm radial margin and a 2-cm craniocaudal margin. In preoperative RT, historically more recent but potentially the most prevalent approach used today, the GTV is treated to a dose of 50 Gy in 25 fractions over 5 weeks with surgery following 4–6 weeks later. In general, the CTV encompasses the GTV with a 4-cm craniocaudal and a 1.5–2.0 cm radial margin for microscopic disease coverage. CTV should also include peritumoral edema as it may harbor tumor cells at some distance from the GTV.91 Following preoperative RT, a postoperative “boost” has traditionally been used but is generally restricted to patients who received preoperative RT and have margin-positive disease at surgery. This is because the local control rate for margin-negative cases is in excess of 90% even when a boost is not provided.81, 92, 93 A positive margin is declared when the tumor reaches the inked surface of the specimen, and clear margins can be declared if the tumor does not reach the ink irrespective of how close it is.81
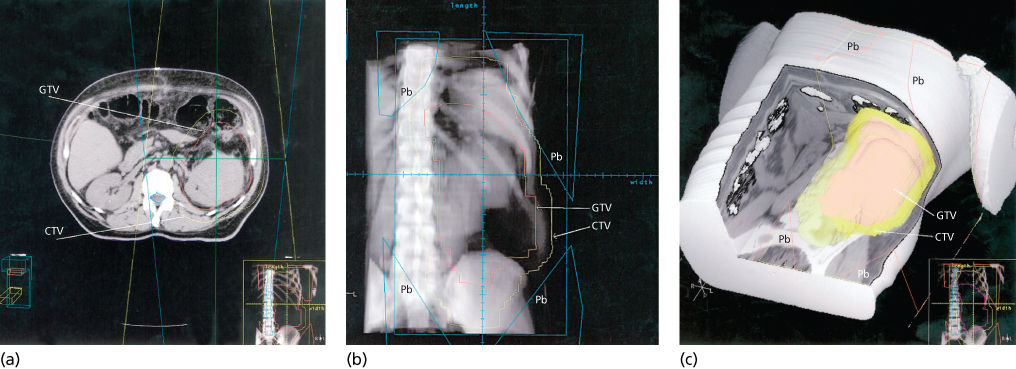
Figure 7 (a) The GTV has been contoured on a CT simulator workstation (red outline). This includes the anterior abnormal fat shown earlier (Figure 5b,c). This process is performed with many thin CT slices to permit reconstruction of the image later for three-dimensional treatment planning. The CTV is outlined in yellow to account for potential microscopic spread beyond the GTV. An additional margin will also be added to account for setup variation and organ motion. Note the displacement of the bowel loops by the tumor mass. The straight lines show the path of the beam for a conventional setup with opposed anterior and posterior fields. (b) The contoured GTV and CTV information displayed in a beam’s eye view (BEV) using a digitally reconstructed radiograph created by the CT simulator. Shielding (Pb) can be placed once the path of the beam within the target areas defined is seen on the BEV. One can also discern the opaque tumor partially displacing the bowel from target area. (c) A three-dimensional reconstruction with the GTV, CTV, and areas to be shielded (Pb) shown with abdominal wall and anterior structures cut away. Generally, these “cut-away” images are most useful for visualizing the edge of the target volume adjacent to critical anatomy that must be protected and when the spatial relationship cannot be verified precisely with conventional imaging.
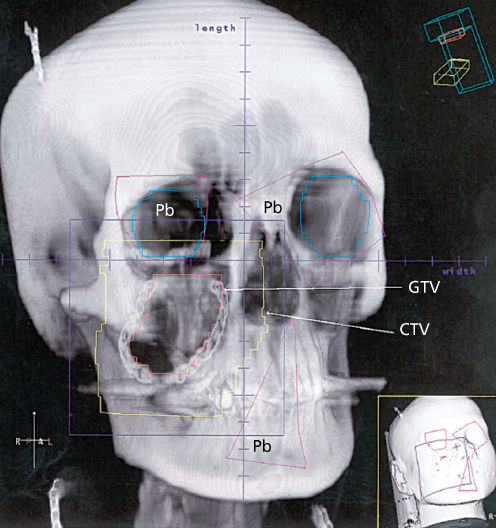
Figure 8 A digitally reconstructed radiograph of the head and neck of a young woman with an STS of the right cheek. Because of the proximity to the right eye, preoperative radiotherapy has been chosen because of its ability to permit maximal restriction of the CTV to the local environment of the tumor. The same process was followed using a CT simulator as described in Figure 6. The GTV on the cheek can be seen with the surrounding CTV. Shielding (Pb) is also evident. A hair clip, which the patient was wearing during the CT slice acquisition, is evident in the right parietal area; one can also see her necklace. The smaller inset shows a three-dimensional image of the patient with potential beams applied.
The need for a radiation therapy boost following preoperative RT and surgery with positive resection margins has been questioned in a retrospective review of 216 ESTS (extremity soft tissue sarcomas) patients; 52 received preoperative RT (50 Gy) alone and were compared to 41 who received preoperative RT with a postoperative boost (generally 16 Gy). A portion of the population did not receive RT at all, or received postoperative RT and were excluded (123 of 216). The postoperative boost cohort had lower 5-year LR-free rates (74% vs 90% for preoperative RT only) indicating that a postoperative boost provides no obvious advantage.94 A similar study (n = 67) yielded almost identical results.95 These results suggest that a benefit from a delayed postoperative boost following preoperative RT and surgery is at best debatable, and the increased risk and challenges of managing later RT morbidity (e.g., radiation-induced fractures) resulting from the higher radiation doses involved should be considered when treating STS.
The defined external beam volumes for extremity STS reflect those of the prospective Canadian Sarcoma Group randomized clinical trial discussed later.96 However, the recently completed UK postoperative phase III randomized “VORTEX” trial (NCT NCT00423618) compared a 5-cm longitudinal margin from GTV in the standard arm to a 2-cm margin and results are awaited.97 A completed RTOG-0630 trial (NCT NCT00589121) addressing the role of IMRT defined slightly smaller preoperative RT volumes (longitudinal 3 cm), although its results may be less easily interpreted owing to its nonrandomized nature.98 In any event, the size of the RT volume margins is not appreciably smaller than the 4-cm longitudinal margin noted above and further outcome reports will be needed in the future to guide practice if target volumes are being reduced in the interest of normal tissue protection.
Despite the variations noted in target volume coverage, the local control rates reported for extremity STS using combinations or surgery and RT are approximately 90% and may suggest that the zone of microscopic involvement may be less than that was previously realized. Recent improvements in surgical technique may lessen the degree of intraoperative tumor dissemination, and irradiation of all surgically handled tissues, scars, and drain sites may be unnecessary. This seems particularly relevant for major centers where surgery is performed by teams with extensive experience in sarcoma management. One must also consider the possibility that case selection factors may explain apparent variations in practice between BRT and external beam RT approaches.
Sequencing of radiotherapy and surgery
The two most common methods of EBRT delivery are preoperative and postoperative RT. Preoperative RT is delivered to an undisturbed and potentially better oxygenated tumor site, which may be one reason why lower preoperative radiation doses do not appear to compromise local control.99 Nielsen et al.100 repeated RT planning in patients who had undergone preoperative RT and surgery and observed that the field size and number of joints irradiated in preoperative RT were significantly less than if the treatment had been administered postoperatively. Another advantage of preoperative RT is that it promotes collaboration between the surgical and radiation oncologists and facilitates the formulation of a coordinated management plan before any treatment.
The Canadian Sarcoma Group SR2 clinical trial represents the only prospective randomized comparison of preoperative versus postoperative RT.96 As was anticipated, the trial showed that preoperative RT results in an increased rate of acute wound complications (WCs). On the other hand, as also anticipated, the trial also showed that postoperative delivery is associated with increased limb fibrosis, edema, joint stiffness, and bone fractures.
Long-term follow-up of patients treated in the Canadian Sarcoma Group NCIC trial (SR2) showed that, of 129 patients evaluable for late toxicity, 48% in the postoperative group compared to 32% in the preoperative group had grade 2 or greater fibrosis (p = 0.07).101 Edema was more frequently seen in the postoperative group (23% vs 16%), as was joint stiffness (23% vs 18%). Patients with these complications had lower function scores (all p values <0.01) on the Toronto Extremity Salvage Score and the Musculoskeletal Tumor Society Rating Scale. Field size predicted greater rates of fibrosis (p = 0.002) and joint stiffness (p = 0.006), and marginally predicted edema (p = 0.06). Acute wound-healing complications were twice as common with preoperative compared to postoperative RT. The increased risk was almost entirely confined to the lower extremity (43% associated with preoperative vs 21% with postoperative timing; p = 0.01). Of interest, additional reports, including one from the University of Texas M.D. Anderson Cancer Center, using the same criteria for classifying WCs as were used in the Canadian NCI trial, found almost identical results.66
The influence of time interval between preoperative EBRT and surgery on the development of WC in extremity sarcoma has also been studied. While the interval had little influence, the data still suggested that the optimal interval to reduce potential WC was 4 or 5 weeks between RT and surgery.67
In the initial report of the SR2 trial with 3.3 years median follow-up, an improvement in overall survival (OS) (p = 0.048) in the preoperative RT arm was noted and partially explained by increased deaths in the postoperative RT unrelated to sarcoma.96 The local failure rate was identical in the 2 arms (7%) (Figure 9). However, updated results were recently presented and the preliminary survival difference had dissipated.101 The 5-year results for preoperative versus postoperative, respectively, were local control, 93% versus 92%; metastatic relapse-free, 67% versus 69%; recurrence-free survival, 58% versus 59%; OS, 73% versus 67% (p = 0.48); and cause-specific survival, 78% versus 73% (p = 0.64). Cox modeling showed only resection margins as significant for local control. Tumor size and grade were the only significant factors for metastatic relapse-free, OS, and cause-specific survival. Grade was the only consistent predictor of recurrence-free survival.
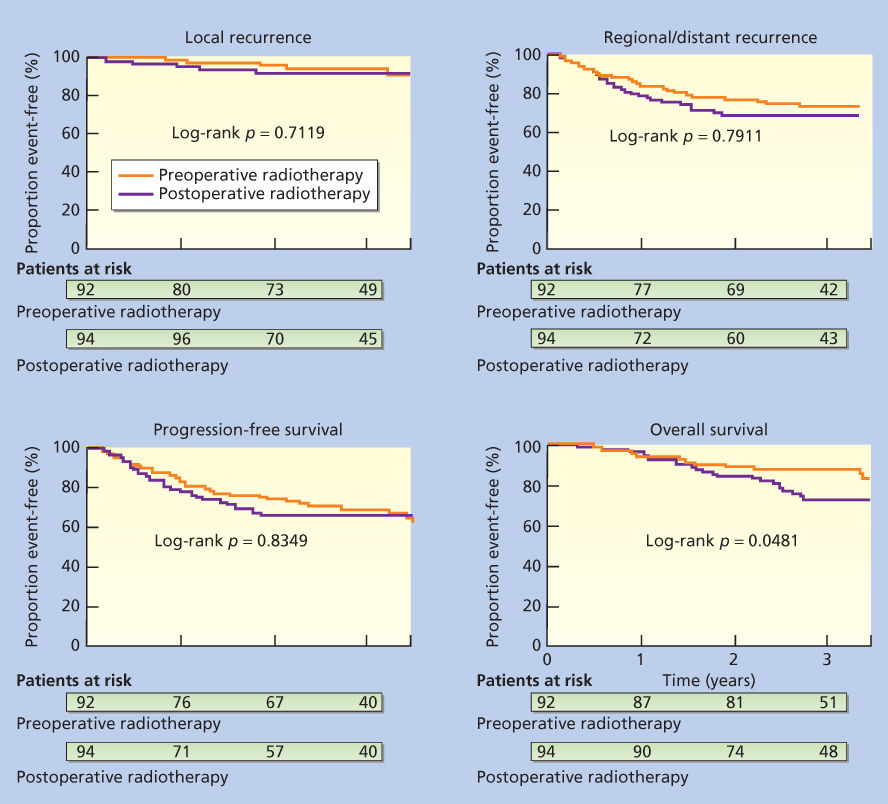
Figure 9 Kaplan–Meier plots for probability of local recurrence, metastasis (local and regional recurrence), progression-free survival, and overall survival in the Canadian Sarcoma Group randomized trial of the NCI of Canada Clinical Trials Group comparing preoperative and postoperative radiotherapy.
Source: O’Sullivan et al. 2002.96 Reproduced with permission of Elsevier.
For the present, decisions about preoperative versus postoperative RT for extremity soft tissue sarcoma should be individualized, taking into account tumor location, tumor size, RT volumes needed, comorbidities, and risks. In general, preoperative RT provides some advantages over postoperative RT, but exposes the patient to significantly increased risks of serious postoperative WCs. A summary of the relative indications that can be used to select patients for preoperative RT is provided in Table 4. In addition, although much of the discussion about preoperative RT is focused on extremity lesions, patients with RPS tolerate preoperative RT substantially better than postoperative RT. This is because the tumor acts as a tissue expander to exclude the bowel from the RT volume (Figure 7a–c). This is discussed in detail later in the section titled “Retroperitoneal Sarcomas.”
Table 4 Relative indications for preoperative RT, despite concerns related to wound complications
| Treatment context/sarcoma site | Issues of concern | Comments |
| Head and neck | ||
| Paranasal sinus | Proximity to optic apparatus (eye, orbit, chiasma) | Major visual functional deficit can be minimized |
| Skull base | Proximity to spinal cord, brain stem | Other “lesser” morbidities (dental, xerostomia) may also be less due to reduced doses and volumes |
| Cheek and face | Xerostomia | Early caries or loss of teeth, loss of sense of taste |
| Split thickness skin graft reconstruction (especially lower limb) | Skin graft breakdown and consequent infection | Many months to years of recreational and/or vocational disability may occur during healing (rare) |
| Large volume GTV or CTV occupying coelomic cavities | ||
| Retroperitoneum | Proximity to the bowel, the liver, and the kidney | Critical organs may be displaced by tumor or not fixed or adherent as is likely in postoperative setting |
| Entire tumor treated before possible contamination of cavity | ||
| Some small bowel lesions | Proximity to critical anatomy, especially intestine with side wall adherence | Contamination of abdominal cavity renders postoperative RT unsuitable |
| Thoracic wall/pleura | Proximity to the lung or the cardiac structures | The lung may be displaced by the chest wall or pleural tumor and can be avoided with preoperative RT, or permits GTV to be treated before operative contamination |
| Abdominal trunk walls, pelvic side wall | Proximity to the kidney, the bowel, the liver, and the ovaries | Avoid CTV encroachment on vulnerable anatomy |
| GTV adjacent to dose limiting critical anatomy | ||
| Thoracic inlet/upper chest | Proximity to brachial plexus | Dose limitation of critical anatomy lends itself to preoperative wall low neck RT. |
| Additional volume considerations | ||
| Medial thigh (young male) | Proximity to testes | Permanent infertility may be avoided |
| Central limb tumor | Proximity to other compartments | Permits partial circumferential sparing, which would not be feasible in postoperative setting |
Abbreviations: CTV, clinical target volume; GTV, gross tumor volume; RT, radiotherapy.
Source: O’Sullivan et al. 1999.77 Reproduced with permission of Elsevier.
Methods of radiotherapy delivery
In general terms, the most generally accepted methods of delivering RT include EBRT and BRT. The former also includes the controversy about its scheduling (preoperative vs postoperative) discussed earlier and in conventional terms also needs attention to the potential role of IMRT that was also mentioned earlier. No randomized trials directly comparing external beam RT and BRT have been undertaken, but both forms of RT have been compared with surgery alone. There also have been no randomized trials addressing IMRT but two prospective phase II trials exist and institutional data are emerging.
Intensity-modulated radiotherapy
Target coverage and protection of normal tissues from high-dose areas appear to be superior for IMRT compared to traditional techniques in ESTS.102 A recent retrospective review spanning noncoincident treatment time periods compared surgery combined with either IMRT (n = 165) or conventional EBRT (n = 154). Allowing for known limitations associated with studies involving treatments deployed over different eras, IMRT showed significantly reduced LR for primary ESTS (7.6% LR for IMRT vs 15.1% LR for conventional RT; p = 0.02).103
Two prospective phase II trials [from Princess Margaret (NCT00188175) and the Radiation Therapy Oncology Group (RTOG 0630: NCT00589121)] investigated if preoperative image-guided radiotherapy (IGRT) using conformal RT/IMRT could reduce RT-related morbidities.98, 104 The characteristics of the Princess Margaret (PMH) and RTOG 0630 trials differed in several ways, particular relating to the exclusion of upper extremity lesions in the PMH trial, the use of a boost following preoperative RT in RTOG 0630 as well as the potential to use chemotherapy in the RTOG trial, and some aspects of the choice of target coverage mentioned below. The two trials also differed regarding their primary endpoints.
The PMH trial showed reduced wound-healing complication (WC) rates (31%) in lower extremity compared to the 43% risk in the previous Canadian Sarcoma Group NCIC SR2 trial that only used 2D and 3D RT.96 The need for tissue transfer, RT chronic morbidities, and subsequent secondary operations for WCs were reduced while maintaining good limb function and local control (93%). The RTOG-0630 trial reported a significant reduction of late toxicities in comparison to the NCIC-SR2 trial (11% vs 37% in SR2), which is very similar to the IGRT PMH trial. Importantly, both the PMH and RTOG-0630 trials defined the CTV differently (longitudinal margin of 3 cm from the gross tumor for high-grade lesions and 2 cm for low-grade lesions versus 4 cm longitudinal margins in the PMH trial). Potentially, the reduction in CTV margins of this degree could explain the improvement in limb function with comparable local control, although an alternative possibility is the reduction in normal tissues receiving the target dose in all dimensions, which is shared by both studies. In the end, it also seems that IMRT is capable of conforming the dose more suitably to the desirable target volume compared to traditional conformal techniques.
Perhaps the greatest advantage to this approach is not only the possibility of improving local control but also the ability to spare bone toxicity and late fractures by achieving bone avoidance, which are often overlooked in the discussion of combined-modality treatments of extremity sarcoma. A recent study addressed evidence-based dose volume bone avoidance objectives for IMRT planning in 230 patients (176 lower and 54 upper extremity) with a median follow-up of 41.2 months.105 The overall risk of fracture was 2% (4/230 patients), which compares favorably to a previous reported incidence of 6%, and suggests that efforts to achieve bone avoidance are appropriate.
Brachytherapy
BRT has some putative advantages over external beam, including a shorter overall treatment time (4–6 days vs 5–6.5 weeks) and quicker initiation of RT after surgery while clonogenic numbers are at a minimum. Because of its brevity, BRT is also more easily integrated into protocols that include systemic chemotherapy than is external beam RT, with its protracted courses. The irradiated volume is also smaller with BRT, which may confer functional advantages. BRT may also have an advantage in situations in which normal tissue tolerance to RT is compromised. One such scenario would be when a postoperative RT boost to the operative bed is desired in patients who received preoperative RT. The use of BRT with surgery in previously irradiated tissues is another situation to achieve limb salvage.106, 107 As noted earlier, no apparent benefit for BRT over surgical excision alone is evident with low-grade lesions, and external beam appears more effective for these tumors (Table 3).69, 70, 108 BRT also permits radiation volumes to be mapped according to intraoperative findings. The American Brachytherapy Society Guidelines differ from those for EBRT and also advise that BRT as a sole treatment modality is contraindicated in the following situations: (1) the CTV cannot be adequately encompassed by the implant geometry, (2) the proximity of critical anatomy, such as neurovascular structures, is anticipated to interfere with meaningful dose administration, (3) the surgical resection margins are positive, and (4) there is skin involved by tumor.109, 110
BRT seems less useful where implant geometry is not optimal, such as in the upper extremity or more proximal limb regions.111 The results of the BRT randomized trial were discussed earlier. In addition, BRT was compared retrospectively to IMRT in 134 high-grade ESTS with similar adverse features.112 The 5-year local control rate was 92% for IMRT compared to 81% for BRT (p = 0.04). Unfortunately, while the results of BRT, including its more restrictive criteria for use, suggest lower efficacy compared to EBRT, there is no randomized controlled trial comparing these seemingly effective local adjuvants.
Traditionally BRT studies, including those mentioned above, used low-dose rate techniques. High-dose rate (HDR) BRT has potential logistic advantages including lower radiation staff exposure, outpatient delivery, and optimized dose distributions by varying dwell times. However, wound-healing complications may occur in sarcoma management and caution is also recommended when placing catheters adjacent to neurovascular structures. As yet, no large series evaluating HDR BRT for STS is available nor has it been directly compared to LDR, partly because of technical differences.
Additional approaches to RT delivery
In addition to external beam RT, BRT, and IMRT, several other approaches for RT delivery exist. These include particle beam RT (electrons, protons, pions, or neutrons), intraoperative radiotherapy (IORT) using external beam or BRT approaches, and combinations of other techniques (e.g., hyperthermia) with RT. IORT has been used most often in the management of RPS and will be discussed later. Some reports also describe IORT for extremity sarcomas.113, 114 Formal clinical trials have not compared the relative merits of these approaches, and their use may be governed as much by the availability of an approach at a given center as by any special advantage that it may confer. In the case of proton beam RT, its ability to achieve accurate targeting provides an advantage when tumors lie in proximity to critical structures.115, 116 In general, however, although reports on the use of many of these approaches exist, the problems of selection bias need to be considered in interpreting these small series in which treatments were not randomly assigned.117, 118
Systemic therapy
Systemic agents, including both traditional cytotoxic chemotherapy drugs and newer small molecule oral kinase inhibitors (SMOKIs), are used widely in the metastatic setting for patients with STSs. The use of chemotherapy in the adjuvant setting remains somewhat controversial. However, if chemotherapy is going to have the same impact as radiation and surgery in the management of sarcomas, more effective drugs must be identified to help improve the cure rate for patients with primary tumors and unseen microscopic metastatic disease. This section will review the use of chemotherapy in the adjuvant and metastatic settings. A brief discussion of chemotherapy combined with radiation therapy is also included in this section.
Adjuvant systemic therapy following primary surgical resection
Although local or local–regional recurrence is a problem for a small subset of patients following primary therapy, the major risk to life in sarcoma patients is uncontrolled systemic disease. The availability of systemic therapy with proven, albeit often limited, ability to induce shrinkage of advanced sarcomas has raised the question of whether the early use of systemic treatment might affect microscopic metastatic disease and yield improvements in OS and disease-free survival (DFS).
Certainly for Ewing sarcoma/PNET, rhabdomyosarcoma, and osteogenic sarcoma, adjuvant or neoadjuvant chemotherapy is an appropriate standard of care.119–122 However, for more common STSs such as leiomyosarcoma, liposarcoma, and high-grade UPS (formerly known as MFH), the benefit of chemotherapy, if there is one, is small.123 As adjuvant therapy is utilized by many practitioners for more common diseases where the benefit is a relatively small one, such as stage I breast cancer and stage II colon cancer, this small potential benefit is an issue that needs to be discussed on an individualized basis. Certainly, the lack of available effective agents for metastatic sarcoma has impeded progress in this area, but the utility of imatinib in both the metastatic and adjuvant setting in GIST gives hope that new agents will contribute to the ultimate goal of any type of systemic therapy specifically increasing the cure rate for people with new diagnoses.
There have been over a dozen studies of anthracycline-based adjuvant chemotherapy for STSs that date back nearly as long as the initial development of doxorubicin.124, 125 These will not be reviewed here, as anthracycline/ifosfamide-based therapy constitutes a better standard of care in patients offered adjuvant chemotherapy, and only one of the studies completed by 1992 had used ifosfamide.
In one of the largest combination chemotherapy studies, the Italian Sarcoma Study Group (ISSG) examined patients with primary or recurrent resected STS of the extremity or limb girdle treated or not treated with radiation.126, 127 A total of 104 patients were randomized to receive no chemotherapy or to receive ifosfamide (1800 mg/m2/day for 5 consecutive days with mesna) and epirubicin (60 mg/m2 on 2 consecutive days), with filgrastim support. Interim analysis in 1996 led to early conclusion of the trial because the study had reached its primary endpoint, specifically improved DFS. At a median follow-up of 36 months, OS in the chemotherapy arm was 72%, compared with 55% for the control arm (p = 0.002). However, with long-term follow-up, the OS difference showed only a trend to statistical significance on an intention-to-treat analysis.126 This study is the strongest single study in the literature supporting the use of adjuvant chemotherapy for STS. Interpretation of the study is made more difficult with the observation of equivalent distant ± LR rates at 4 years.
Conversely, no survival benefit was observed in an EORTC phase III study of adjuvant chemotherapy versus observation (doxorubicin 75 mg/m2, ifosfamide 5 g/m2 per cycle for 5 cycles, with filgrastim support).128 A total of 351 patients were accrued between 1995 and 2003, and 130 (80%) of the 163 patients receiving chemotherapy completed all 5 cycles. OS was not statistically different between arms (HR 0.94, 95% confidence interval 0.68–1.31 for the treatment arm, p = 0.72) and relapse-free survival was also not statistically different (HR 0.91, CI 0.67–1.22, p = 0.51). The 5-year OS rate was 67% for the treatment arm and 68% for the control group. The major differences between this study and the ISG study were a lower dose of ifosfamide and use of epirubicin in the ISSG trial, but it is unclear based on data from metastatic disease of the relevance of these differences.
The most recent and comprehensive overview of adjuvant chemotherapy for extremity sarcomas to date was the 2008 meta-analysis of 18 studies encompassing sarcomas of all anatomic sites.129 In this analysis, 93 potential studies were considered and 18 ultimately selected, constituting 1953 patients with STSs of extremity and nonextremity. Pathology review was not centralized. The results of the meta-analysis, including the actuarial outcome probabilities and the hazard ratios, are summarized in Figure 10 and Table 5.
Figure 10 Actuarial curves from individual patient data meta-analysis: (a) local recurrence-free interval (RFI), (b) distant recurrence-free interval, (c) overall recurrence-free survival, and (d) overall survival.
Source: O’Bryan et al. 1977.130 Reproduced with permission of Wiley.
Table 5 Relative risks and 95% confidence intervals for clinical outcomes with adjuvant chemotherapy (2008 meta-analysis)
| Doxorubicin | Doxorubicin–ifosfamide | Combined | |
| Local RFI | 0.75 (0.56–1.01) | 0.66 (0.39–1.12) | 0.73 (0.56–0.94) |
| Distant RFI | 0.69 (0.56–0.86) | 0.61 (0.41–0.92) | 0.67 (0.56–0.82) |
| Overall RFS | 0.69 (0.56–0.86) | 0.61 (0.41–0.92) | 0.67 (0.56–0.82) |
| Overall survival | 0.84 (0.68–1.03) | 0.56 (0.36–0.85) | 0.77 (0.64–0.93) |
Source: Pervaiz et al. 2008.129 Reproduced with permission of Wiley.
Study data from the 18 trials were combined without examining individual patient data. Combining all data, LR risk, distant recurrence risk, overall recurrence risk, and OS were superior with chemotherapy. For OS, the relative risk of death was 0.77 with chemotherapy (95% CI, 0.64–0.93), p = 0.01, and relative recurrence risk was 0.67 (95% CI, 0.56–0.82), p = 0.0001. The absolute risk reduction of any recurrence was 10% and absolute risk reduction for OS was 6%. However, as pointed out in a commentary on a prior meta-analyses,131 these data have to be interpreted with caution. For example, (1) individual patient data were not reviewed and interrogated, (2) in an older analysis, 18% of patients did not have histology available for review, recognizing the error rate in pathology review at expert centers, (3) ineligibility rates were high, and (4) the largest individual trial was not included (published after the meta-analysis publication). Although the meta-analysis cannot replace a well-designed randomized study, it reinforces many of the findings from smaller studies that local and distant recurrence-free survival is definitely improved, and OS modestly so, at least in unselected patients.
Preoperative (neoadjuvant) chemotherapy
Preoperative chemotherapy has theoretical advantages over postoperative treatment. First, preoperative chemotherapy provides an in vivo test of chemotherapy sensitivity. Patients whose tumors show objective evidence of response are presumed to be the subset that will benefit most from further postoperative systemic treatment. In contrast, it is assumed that the population of nonresponding patients will derive minimal or no benefit from further chemotherapy and can therefore be spared its toxicity. On the other hand, it is conceivable that the patients whose tumors respond to chemotherapy may not be those who would derive the most from chemotherapy, because these lesions with favorable biology might be those destined to do well irrespective of any systemic treatment. In contrast, those who do not respond may be those with unfavorable disease who could derive the greatest benefit from the discovery of highly effective systemic treatments.
A second potential advantage of preoperative chemotherapy is that it treats occult microscopic metastatic disease as soon as possible after the cancer diagnosis. This may theoretically prevent the development of chemotherapy resistance by isolated clones of metastatic cells or prevent the postoperative growth of microscopic metastases, but given the nature of growth of sarcomas, at most one or two doublings of the tumor would be affected, far fewer than the >35 typically required in the development of a >1-cm tumor. Chemotherapy-induced cytoreduction may permit a less radical and consequently less morbid surgical resection than would have been required initially. In patients with large STS of the extremities, cytoreduction may reduce the morbidity of limb-sparing surgical procedures and possibly even allow patients who might otherwise have required an amputation to undergo limb-sparing surgery.
Investigators from the MD Anderson Cancer Center reported long-term results with doxorubicin-based preoperative chemotherapy for AJCC stages IIC and III (formerly AJCC stage IIIB) extremity STS.132 In a series of 76 patients treated with doxorubicin-based preoperative chemotherapy, radiologic response rates included complete response, 9%; partial response, 19%; minor response, 13%; stable disease, 30%; and disease progression, 30%. The overall objective major response rate (complete plus partial responses) was 27%. At a median follow-up of 85 months, 5-year actuarial rates of LR-free survival, distant metastasis-free survival, DFS, and OS were 83%, 52%, 46%, and 59%, respectively. The event-free outcomes reported from MD Anderson are similar to those observed with chemotherapy in the phase 3 postoperative chemotherapy trials. Furthermore, comparison of responding patients (complete and partial responses) and nonresponding patients did not reveal any significant differences in event-free outcome. Conversely, only 1/29 patients in a smaller study from Memorial Sloan-Kettering demonstrated WHO defined tumor shrinking after two cycles of doxorubicin-based therapy.133
Ifosfamide-containing combinations also have been used in the preoperative setting. Selected patients treated with aggressive ifosfamide-based regimens have had major responses, and preliminary results suggest that response rates may be higher than in historic controls treated with non-ifosfamide-containing regimens.134 However, as noted above, the randomized phase 2 neoadjuvant study of doxorubicin and ifosfamide chemotherapy showed no benefit for the treatment arm, although the study was not specifically designed to determine a survival advantage.135
Regional administration of chemotherapy
It is hypothesized that the antineoplastic action of adjuvant chemotherapy will be improved by modifying factors related to drug delivery. One such modification is administering chemotherapy regionally rather than systemically. Intraperitoneal chemotherapy is used in primary therapy of ovarian cancer or appendiceal carcinoma, where it remains more superficial and accessible to peritoneal chemotherapy compared to other cancers that more commonly form more distinct masses that intraperitoneal chemotherapy cannot penetrate. For a period of time, intraperitoneal chemotherapy was used for GIST and gastrointestinal leiomyosarcomas that spread to the peritoneum, but this approach was abandoned given the access to tyrosine kinase inhibitors for GIST. Perhaps the situation in which regional therapy is most commonly used for sarcoma is intra-arterial chemotherapy used in some centers for osteogenic sarcoma, which provides both a higher concentration of drug locally as well as systemic effects after infusion. In some situations, chemotherapy is administered with radiation (see below).
Other investigators evaluated whole-body or regional hyperthermia to enhance the efficacy of combination chemotherapy, using methods to increase the temperature of the entire body or a specific region alone to 41°C.136–138 In the 341 patient randomized study of primary and locally recurrent high-risk sarcomas, combination of preoperative hyperthermia and EIA chemotherapy (etoposide, ifosfamide, and doxorubicin) was superior to EIA alone in terms of local control and disease-free survival, although OS was not impacted. The study was criticized for not being able to sort out the contribution of radiation and hyperthermia, and what appeared to be a high frequency of R1 and R2 resections for patients going on study. Concurrent hyperthermia and chemotherapy is an approved treatment in some European countries, but remains investigational in the United States.
Combined preoperative chemotherapy and RT
Data regarding combination chemotherapy and radiation are more developed than hyperthermia and chemotherapy, largely from the commonality of the former being available at various institutions. As with combinations with hyperthermia, the primary putative advantage of preoperative chemotherapy with radiation is the potential to shrink selected lesions resectable only by amputation sufficiently that they become amenable to a limb-sparing approach.
Concurrent chemotherapy and radiation has been employed extensively by Eilber and colleagues at UCLA and has been modified and examined by other groups.139, 140 The first chemotherapy–radiation treatment protocol typically involved intra-arterial doxorubicin with high dose per fraction RT (35 Gy of external beam radiation delivered in 10 daily fractions, which was reduced to 17.5 Gy in 5 daily fractions to minimize local toxicity). Although the intra-arterial route delivers chemotherapy more directly to the tumor, it is more complex, expensive, and prone to complications than intravenous chemotherapy.141 Indeed, a prospective randomized trial comparing preoperative intra-arterial doxorubicin with intravenous doxorubicin, both followed by 28 Gy of radiation delivered over 8 days and then surgical resection, showed no differences in LR or survival.140
The largest study to date directly studying systemic chemotherapy combined with RT examined razoxane as the radiation-sensitizing agent in a randomized study of drug versus no drug in combination with radiation therapy for resectable or unresectable STSs.142 Acute skin reactions were enhanced in the razoxane arm, but late toxicity was not greater than in the control arm. Although there are imbalances in the arms of the study, for the 82 of 130 evaluable cases examined with gross disease RT (median dose 56–58 Gy) with razoxane (daily oral doses of 150 mg/m2 throughout RT) showed an increased response rate (74% vs 49%) and improved local control rate (64% vs 30%; p < 0.05) compared with external beam radiation alone.
Either ifosfamide or cyclophosphamide has been routinely combined with radiation therapy as part of the definitive therapy for Ewing sarcoma and rhabdomyosarcoma in an attempt to continue systemic therapy at the same time as maximizing local control.119 In general, toxicity does not appear to be greater than that seen for radiation alone. However, skin toxicity from the combination was greater in one study than that seen with radiation alone.143
An alternative sequential chemotherapy and radiation strategy in patients with localized, high-grade, large (>8 cm) extremity STSs has been examined.144–146 This treatment protocol involved 3 courses of doxorubicin, ifosfamide, mesna, and dacarbazine (MAID) with two 22-Gy courses of radiation (11 fractions each) for a total preoperative radiation dose of 44 Gy. This was followed by surgical resection with careful microscopic assessment of surgical margins. An additional 16-Gy (8 fraction) boost was delivered for microscopically positive surgical margins. The outcomes of 48 patients treated with this regimen were compared with those of matched historic controls and was superior to that of the historical control patients.144 The 5-year actuarial local control, freedom from distant metastasis, DFS, and OS rate were 92% versus 86% (p = 0.1155), 75% versus 44% (p = 0.0016), 70% versus 42% (p = 0.0002), and 87% versus 58% (p = 0.0003) for the MAID and control patient groups, respectively. Febrile neutropenia was a complication in 25% of patients. Wound-healing complications were substantial and occurred in 14 (29%) patients receiving the chemotherapy/radiation sequential therapy. One patient who received chemotherapy developed late fatal myelodysplasia. Given the favorable results of this study in comparison to historical controls for high-risk extremity STS, the Radiation Therapy Oncology Group conducted a multi-institutional trial, modifying the chemotherapy in an attempt to address the local toxicity issue. The report of the trial suggested combined-modality treatment can be delivered successfully in a multi-institutional setting albeit with some toxicity. Efficacy results are consistent with previous single-institution results.145, 146 The question remains whether this approach in high-risk, extremity STS confers significant survival benefits following and intense regimen of neoadjuvant chemoradiotherapy and surgery, which seems sustained even with long-term follow-up.
Although significant toxicity was observed, local control was improved in the prior studies compared to historical controls, raising the possibility that combined chemotherapy and radiation could be combined safely to decrease local control risk. Pisters et al.132 examined concurrent doxorubicin and irradiation in the neoadjuvant setting in 27 patients with extremity STS. Preoperative external beam radiation was administered in 25 fractions of 2 Gy each. Doxorubicin was administered in escalating doses with a bolus followed by 4-day continuous infusion weekly. Radiographic restaging was performed 4–7 weeks after chemoradiation. Patients with localized disease underwent surgical resection. The maximum tolerated dose of continuous infusion doxorubicin combined with standard preoperative radiation was 17.5 mg/m2/week; 7 of 23 (30%) patients had grade 3 dermatologic toxicity at this dose level. Macroscopically complete resection (R0 or R1) was performed in all 26 patients who underwent surgery. In 22 patients who were treated with doxorubicin at the maximum tolerated dose and subsequent surgery, an encouraging 11 patients (50%) had 90% or greater tumor necrosis, including 2 patients who had complete pathologic responses. This approach appears valid with other radiation-sensitizing agents as well, such as gemcitabine.147 Further studies of combination therapy are also discussed later in the section titled “Retroperitoneal Sarcomas.”
Other multicenter trials of adjuvant therapy for STS
It is well recognized that different sarcoma subtypes have different chemotherapy resistance/sensitivity patterns, details of which are discussed in greater detail elsewhere.61 For example, synovial sarcoma is typically resistant to gemcitabine–docetaxel, and leiomyosarcomas are typically less sensitive to ifosfamide than other forms of sarcoma. Synovial sarcoma and myxoid/round-cell liposarcoma appear to be more sensitive to chemotherapy in the metastatic setting than other subtypes of sarcoma and may well be two subtypes that respond to both anthracyclines and ifosfamide. These data argue that adjuvant chemotherapy should be examined on a subtype-specific basis. Combined nonrandomized data from UCLA and MSKCC showed that adjuvant chemotherapy may indeed be useful in the setting of synovial sarcomas and myxoid/round-cell liposarcoma and argued for the use of chemotherapy in the neoadjuvant setting for all types of STS, based on institutional databases.148–150 Notably, data of all patients treated or not treated with chemotherapy from MD Anderson and MSKCC indicated in the adjuvant or neoadjuvant setting that there was no statistical difference in OS in the group of patients who received chemotherapy versus those who did not.151 However, these data are inherently biased in that it is likely that younger, healthier patients with higher risk tumors were those selected to receive chemotherapy. Even though there was no statistically significant difference between the group of patients who received chemotherapy and those who did not, there were still shifts in the frequency of patients with larger sarcomas with a predominance of liposarcoma toward receiving chemotherapy, perhaps representing the selection bias that allowed a group of patients with an inherently poorer outcome to do as well as those with a better outcome.
In summary, for AJCC stage III STS, if there is a benefit to chemotherapy in the adjuvant setting, it appears to be a modest one. There is variation in practice between centers and between practitioners as to who is an appropriate candidate for chemotherapy. Given this situation, it is the authors’ practice to attempt to compare benefits and risks of systemic therapy and individualize the plan of treatment for a given clinical setting. Patients below age 50 may be those who benefit most among this very heterogeneous patient population. Certainly, the finding of 35% or more of STS with specific genetic translocations or mutations brings hope that the benefit seen with GIST and adjuvant imatinib therapy will carry over to the adjuvant setting for a subset of patients with STS of the extremities and trunk when traditional adjuvant cytotoxic chemotherapy is combined with novel therapeutics.
“Pediatric” sarcomas and adjuvant therapy
The standard of care for nearly all sarcomas specific to the pediatric population involves chemotherapy. It is of proved benefit in patients with osteogenic sarcoma, Ewing sarcoma, and rhabdomyosarcoma. A few brief comments on these chemotherapy-responsive tumors follow.
Neoadjuvant chemotherapy is the standard of care for the initial treatment of osteogenic sarcoma.121 In the era before systemic therapy, cure rates for osteogenic sarcoma, even in the setting of amputation for primary disease, was only on the order of 15%. With chemotherapy, the survival rate is 65–70%. Yet the 30–35% of patients who relapse remain a frustrating problem because the addition of new agents has changed the chance for cure comparatively modestly, and the one agent demonstrating benefit in a randomized trial was not approved in the United States.120 The outcome of adjuvant chemotherapy (degree of tumor necrosis after chemotherapy) is directly associated with improved clinical outcome and provides the opportunity for changing chemotherapy for a poor initial response. The standard of care for adjuvant chemotherapy is a backbone of cisplatin and doxorubicin, with methotrexate employed in most pediatric and some adult patients. The benefit for methotrexate as part of adjuvant treatment was called into question in a 1997 paper in which the combination of doxorubicin and cisplatin alone was shown equivalent to a more complicated and toxic regimen containing high-dose methotrexate.152 However, the methotrexate component of neoadjuvant therapy has been observed to be important in a variety of studies and remains an integral part of combination therapy for osteosarcoma.153 Less questionable is the benefit from nonspecific immune system stimulator muramyl tripeptide (MTP), which was shown in a large pediatric clinical trial to improve OS when used in the adjuvant setting, while there was no benefit in survival seen with the addition of ifosfamide to the standard methotrexate–doxorubicin–cisplatin (MAP) backbone.120
Through a series of international clinical studies, the adjuvant program for rhabdomyosarcoma typically involves an induction course of chemotherapy, followed by combination chemotherapy and radiation, followed by the completion of chemotherapy, which will last approximately 48 weeks in pediatric population. The standard of care is the combination of vincristine, dactinomycin, and cyclophosphamide, as this combination was shown as effective as VAI and VIE and less toxic.119 The addition of doxorubicin to the vincristine, dactinomycin, and cyclophosphamide regimen did not appear to improve OS and is omitted in the treatment of pediatric rhabdomyosarcoma.154 The frequent dosing of vincristine is extremely difficult to complete for adults and requires dose adjustment or shorter courses of therapy for adults than for children with this diagnosis. Children with this diagnosis appear to fare better than adults with the same diagnosis stage in most studies, as well as in everyday practice.155, 156
For patients with Ewing sarcoma, in distinction from rhabdomyosarcoma, the addition of additional agents (ifosfamide and etoposide) to an existing backbone of vincristine, doxorubicin, and cyclophosphamide chemotherapy improved outcome for localized disease, with a 2-week schedule superior to a 3-week schedule of treatment;122 however, not published were data that the 2-week schedule was not beneficial for patients over age 18. This 5-drug regimen is a good standard of care for patients with a new diagnosis of Ewing sarcoma. It is often difficult to administer the 14 cycles of chemotherapy to adults, and abbreviation to the adjuvant therapy program is often necessary. It is also not clear if all 14 cycles are necessary for best outcomes. As with rhabdomyosarcoma, children with Ewing sarcoma fare better than adults with the same diagnosis, all else being equal.157–159
Treatment of locally advanced disease
Hyperthermic isolated limb perfusion, isolated limb infusion, and regional hyperthermia
Hyperthermic isolated limb perfusion (ILP), an investigational technique in the United States (although recently approved by regulatory agencies in other parts of the world), has received considerable attention in the treatment of locally advanced, unresectable sarcomas of nonosseous tissues. ILP involves local perfusion of high-dose chemotherapy (most commonly melphalan) and, when available, tumor necrosis factor alpha (TNFα) under hyperthermic conditions. An oxygenated circuit is established by local arterial and venous cannulation on a bypass pump. Systemic circulation is minimized by placement of a tourniquet proximally.
ILP has been evaluated in two settings: (1) attempted limb preservation in cases of locally advanced extremity lesions surgically amenable only to amputation and (2) function extremity preservation for the short survival duration anticipated in cases of locally advanced extremity lesions with synchronous pulmonary metastases (stage IV disease).
A multicenter phase 2 trial evaluated a series of 55 patients with radiologically unresectable extremity STS using HILP with high-dose TNFα and melphalan, and interferon-α in some patients. 160 A major tumor response was seen and limb salvaged in over 80% of patients. Regional toxicity was limited, and systemic toxicity was minimal to moderate. There were no treatment-related deaths. Despite the high rate of complete responses (15–30%) and limb sparing (>80%) achieved by ILP, no randomized trials have compared ILP to aggressive limb-sparing resection with RT for STS. Therefore, ILP should be considered as a potential treatment, when other options are limited or not available, in appropriately selected patients. Eligible patients should be referred to centers where this therapy is available.
Isolated limb infusion (ILI) has been evaluated in extremity STS in a more limited manner, and parallels work done with intra-arterial chemotherapy. Like IPL, ILI relies on circulating high-dose chemotherapy in an isolated extremity. Unlike ILP, ILI is conducted through percutaneously placed catheters and is performed under hypoxic conditions. However, there is limited experience with ILI in extremity STS. Similar to ILP, ILI has not been directly compared to aggressive limb-sparing resection with EBRT in a randomized trial. However, patients under consideration for ILP and ILI are often no candidates for surgery with EBRT at first evaluation, and therefore ILP and ILI may be considered as potential therapies in appropriately selected patients.
Definitive radiation for local control
Apart from patients with some very radiosensitive subtypes of sarcomas, most patients who undergo RT as the sole treatment modality for sarcoma have been deemed to have locally advanced unresectable disease. RT alone is a rare treatment choice that should be done only at centers skilled in the management of sarcomas; medically fit patients with grossly “unresectable” but nonmetastatic disease should always be referred to a specialty center for multidisciplinary management, which may combine surgery, RT, and possibly chemotherapy. For example, proximal inguinal or axillary tumors that encircle major vascular structures in the proximal leg or arm may be resected along with the involved vasculature and the vessels reconstructed. Adjuvant RT is also generally used. Rarely, a patient with truly inoperable locally advanced disease may require RT alone, with either photon or particle (proton, neutron, or pion) beams.161–163 No formal clinical trials have been performed to compare these strategies with each other, and they are generally administered in an adverse clinical setting. Local control has been reported in 40–70% range.
Treatment of metastatic disease
Clinical problem of metastases
The diagnosis of recurrent or metastatic disease in patients with STSs is often heartbreaking. Patients and physicians are aware that, in general, such a diagnosis is typically fatal. The role of the multidisciplinary sarcoma team in the management of patients with metastatic sarcoma is to recognize opportunities in which multimodality care might still improve important outcomes such as survival or quality of life. Both surgery and systemic chemotherapy can play an important role in improving these outcomes in selected patients. Overall, it is important to recognize that chemotherapy is usually given with the palliative aim of prolonging life and improving quality of life.
The most common site of metastasis from STS of the extremities is the lungs. Indeed, the lungs are the only site of metastasis in approximately 80% of patients with metastases from primary extremity and trunk STS.61 Primary visceral and gastrointestinal sarcomas such as GIST commonly metastasize to the liver, while other visceral sarcomas may metastasize to lungs as well. Extrapulmonary metastases are uncommon forms of first metastasis from extremity sarcomas and usually occur as a late manifestation of widely disseminated disease. The median survival after development of distant metastases is approximately 12 months (Figure 11),165 although more contemporary data suggest that median survival may presently be 15–18 months for unselected patients who receive cytotoxic chemotherapy; the optimal treatment of patients with metastatic STS requires an understanding of the natural history of the disease and individualized selection of treatment options based on patient factors, disease factors, and limitations imposed by prior treatment.
Figure 11 Postmetastasis survival (from time of diagnosis of M1 disease) in a cohort of 230 patients with primary STS of the extremities. The median postmetastasis survival was 11.6 months.
Source: Slater et al. 1986.164 Reproduced with permission of Elsevier.
The approach to patients with advanced or metastatic sarcomas is changing over time. We increasingly realize clinical trials must be stratified rationally for data of value to be derived. Studies of “sarcomas” without stratification by histology or molecular features will soon seem as naïve as studies of “cancer” without further qualification. These mesenchymally derived diseases lumped under the heading of “sarcomas” can be quite different, and studies need to take that into account. In a similar manner, the consideration of a specific sarcoma histology may be supplemented or even superseded by the genetic profile of the tumor. To generate studies of sufficient size and power, large-scale collaborations on a national and international level will be required. Such collaborations are already in place among the nations of Europe individually and collectively (e.g., the Soft Tissue and Bone Sarcoma Group of the EORTC), Canadian centers, and cooperative trials groups and other clinical trials collaborations such as SARC (Sarcoma Alliance for Research through Collaboration) in the United States. With these collaborations, it is hoped that further research will rapidly translate research findings into the novel therapeutics that are so desperately required by patients with sarcomas.
Resection of metastatic disease
Many investigators have reported their experience with pulmonary metastasectomy for metastatic STS in adults.166, 167 Three-year survival rates following thoracotomy for pulmonary metastasectomy range from 23% to 54%. As a result, some patients can be resected with curative intent, similar to the situation for osteogenic sarcoma. As the ability to completely resect all metastatic disease is an important determinant of outcome, the reported interinstitutional variability in postmetastasectomy survival rates is partially a function of whether survival was reported for all patients who underwent thoracotomy or only for the subset who underwent complete resection.
It remains difficult to predict which patients with pulmonary metastases will benefit from pulmonary resection. A number of clinical criteria have been evaluated by univariate analysis in this regard, including the disease-free interval, number of metastatic nodules, and tumor doubling time. Multivariate analyses from both the NCI and Roswell Park Cancer Institute confirm that a short disease-free interval and incomplete pulmonary resection are adverse prognostic factors for survival for patients with pulmonary metastases.166–169 A multivariate analysis from MD Anderson suggested that, in addition, the presence of more than three metastatic pulmonary nodules on preoperative chest CT is an adverse prognostic sign. The most important prognostic factor impacting survival appears to be the ability to completely resect all disease. In the review of postmetastasectomy outcomes in one series, the median survival among patients who were able to undergo complete resection of metastases was 20 months as compared with 10 months among patients who did not have complete resection (Figure 12).165 In summary, the ability to achieve complete resection and the number of pulmonary nodules present appear to best define the postoperative prognosis for these patients.
Figure 12 Postmetastasis survival stratified by resection of pulmonary metastatic disease. The median survival among patients undergoing complete resection of pulmonary metastatic disease was 20 months.
Source: Billingsley et al. 1999.165 Reproduced with permission of Wiley.
Unfortunately, metastasectomy benefits only a fraction of patients who develop pulmonary metastases. This is best illustrated by data from MSKCC, where a population of 716 patients who presented with primary extremity sarcoma were followed for the subsequent development and treatment of pulmonary metastases (Figure 13).170 Of an initial group of 716 patients, 148 patients (21%) developed pulmonary metastases. Isolated pulmonary metastases occurred in 135 (91%) of these 148 patients. Of the 135 patients with pulmonary-only metastases, 78 (58%) were considered to have operable disease, and 65 (83%) of those taken to thoracotomy were able to undergo complete resection of all of their pulmonary metastatic disease. Thus, 44% of all patients with pulmonary metastases were able to undergo complete metastasectomy. The median survival from the time of complete resection was 19 months, and the 3-year survival rate was 23%. All patients who did not undergo thoracotomy died within 3 years. For the entire cohort of 135 patients developing pulmonary-only metastases, the 3-year survival rate was only 11%.
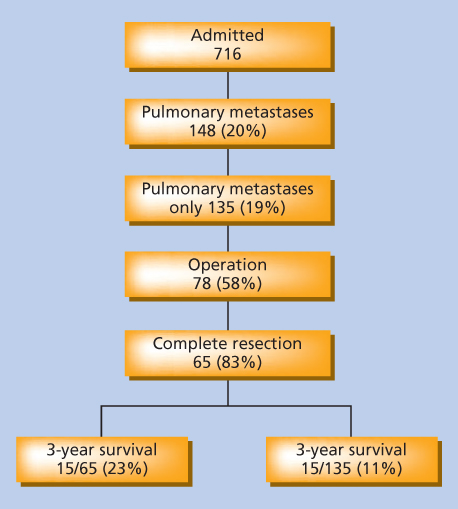
Figure 13 Risk and subsequent management of pulmonary metastases in 716 patients with primary or locally recurrent extremity STS.
Source: Brennan 1996.170 Reproduced with permission of Elsevier.
Several series of repeat pulmonary metastasectomy also have been published. In a series of 43 patients thus treated at the NCI, 72% of patients could be rendered free of disease at the second thoracotomy, with a median survival duration from the time of second thoracotomy of 25 months.171 In a report from MD Anderson of a series of 39 patients undergoing reoperation for a second pulmonary metastasis after successful initial metastasectomy, factors predicting long-term survival included the presence of a solitary metastasis and the ability to perform a complete resection.172 Patients with isolated second metastatic sites fared better than those with multiple resected lesions.
The disappointing overall results of treatment of metastatic disease underscore the importance of careful patient selection for resection of pulmonary metastases. The following criteria are generally agreed upon: (1) the primary tumor is controlled or is controllable, (2) there is no extrathoracic disease, (3) the patient is a medical candidate for thoracotomy and pulmonary resection, and (4) complete resection of all disease appears possible. With careful patient selection, the morbidity of thoracotomy (or repeated thoracotomies) can be limited to the subset of patients who are most likely to benefit from this aggressive treatment approach. The potential role of systemic adjuvant chemotherapy following complete metastasectomy is discussed below in the section “Individualized Therapy” on individualizing chemotherapy for metastases.
Chemotherapy for metastatic disease
Natural history of metastases
A good place to begin a discussion of chemotherapy for unresectable metastatic sarcoma is the expected course of the disease. The EORTC contributed greatly to define the expected course of unresectable metastatic sarcoma by publishing its large series of more than 2000 patients with advanced sarcomas of soft tissues to describe prognostic features and the response to anthracycline-based chemotherapy in an era before GIST was recognized as a unique entity.173 In this study reviewing more than 20 years of experience, the median OS was approximately 1 year. Subsets of patients had longer median survival; such patients were typically those who were younger, had a better performance status, had low-grade sarcoma, had no liver metastases, and had developed metastatic disease following a longer interval from initial diagnosis. Importantly, this study concluded that the variables predicting improved survival were actually different from variables predicting objective response to chemotherapy (the latter variables include such items as high-grade tumor and liposarcoma subtype). Thus, one interpretation that is reasonable is that the most important predictors of survival with metastatic sarcoma are variables dependent on the tumor biology itself, as well as certain patient factors such as age and comorbid disease. These data are critical to understand so that information regarding the impact of new drugs and treatments can be interpreted appropriately, based on a comparison with the correct expectations for the natural or treated history of the disease in past clinical trials.
Individualized therapy
The approach to patients with advanced or metastatic sarcomas is evolving as are new therapeutics, and in this context, the use of therapies directed at specific histologies or DNA alterations is evolving, as are the entirely orthogonal approaches involving immunotherapy. We increasingly recognize that clinical trials must be stratified rationally for data of value to be derived. Studies of “sarcomas” without stratification will soon seem as naïve as studies of “cancer” without further qualification; each of the >50 mesenchymal cancers lumped under the heading “STSs” has a distinct biological behavior, and studies need to take that into account. To generate studies of sufficient size and power, large-scale collaborations on a national and international level will be required.
Tremendous effort has gone into testing multiple, commercially available and experimental agents in STS. Of all of these tested since the common use of ifosfamide, only trabectedin has been approved in Europe and pazopanib in many countries for use in metastatic soft tissue sarcoma; eribulin was also recently approved in the U.S. for liposarcoma; behind this are other novel variations on standard agents being studied, such as aldoxorubicin. For specific diagnoses, imatinib and other SMOKIs have activity, but are not specifically approved for use in sarcomas, save for imatinib in DFSPs. That said, gemcitabine with either docetaxel or DTIC in combination has been shown superior in progression-free and overall survival versus gemcitabine only in randomized studies. Some highlights of these data are noted in the following section.
Anthracyclines and ifosfamide
With the caveat that specific sarcomas demonstrate differential sensitivity to different chemotherapy agents, doxorubicin and ifosfamide remain the most active agents for metastatic sarcoma, with RECIST response rates in the 10–20% range for each drug.174 Depending on the ratio of sensitive versus less sensitive subtypes of STSs in past studies, the response rate can be significantly higher. For example, synovial sarcoma and myxoid/round-cell liposarcomas are relatively sensitive to ifosfamide (as well as doxorubicin), while GIST, alveolar soft part sarcoma, and extraskeletal myxoid chondrosarcoma appear to be largely resistant to both agents.
It is important to recognize that response rates per se increasingly are being criticized as poor surrogates of clinical benefit. Many sarcomas contain acellular desmoplastic stromal tissues. Even when chemotherapy successfully induces significant tumor cell kill in vivo, the matrix left behind appears largely unaffected, leading to falsely negative imaging findings of tumor response to chemotherapy. Thus, objective response rates based on imaging may underestimate the antitumor efficacy of chemotherapy. Conversely, simply shrinking a tumor and achieving a nondurable response may not be worth the toxicities of aggressive multiagent chemotherapy. Thus, from both standpoints, RECIST-defined responses may not be an ideal indicator of antitumor efficacy in sarcoma management in general, with GIST a case in point.175, 176 Increasing attention in the field of sarcoma drug development is thus being paid to other important indicators of clinical outcomes, such as progression-free survival duration, percentage survival at a given time point, and OS rate.
Some drugs may slow disease progression and prolong survival even if objective response rates are low, although the clinical data to support those claims must be generated with rigor and careful attention to consistency of follow-up. Nonetheless, despite observations that clinical benefit might be underestimated by RECIST, it remains the yardstick by which radiological responses are measured.
Dose–response relationships
The sensitivity of sarcomas to chemotherapy was first convincingly demonstrated with doxorubicin in the early to mid 1970s.124 Subsequent studies of doxorubicin in sarcomas have widely been viewed as supporting a dose–response relationship, with doses of 50 mg/m2/cycle or less associated with less antitumor activity than doses of 60 mg/m2/cycle or higher. Although a dose–response relationship is evident, it is important to recognize that other variables may affect antitumor efficacy, such as histopathologic subtype of sarcoma, as noted above. Nonetheless, as a dose threshold for optimal activity has been documented with doxorubicin in another chemotherapy-sensitive solid tumor, specifically breast cancer, it seems reasonable to conclude that doxorubicin is best used at doses above 60 mg/m2/cycle. In addition, analogous to breast cancer, improved response rates above 75 mg/m2/cycle dose range are difficult to demonstrate.
A wide variety of dose- and schedule-ranging studies have been performed with ifosfamide. It is clear that antitumor response is improved by higher doses of ifosfamide.134, 177 This point has been made most convincingly by the responses to high-dose ifosfamide (≥10,000 mg/m2/cycle) in patients who had previously failed to respond to the same drug at lower doses (i.e., ≤6000 mg/m2/cycle). However, given the toxicities of this drug at higher doses, high-dose ifosfamide is best reserved for a subset of patients with disease that is expected to be chemotherapy sensitive to achieve meaningful responses (e.g., before planned surgical extirpation of metastases).
Single-agent versus combination chemotherapy
A continuing controversy is whether the optimal approach to patients with advanced sarcomas is combination chemotherapy regimens or sequential single agents. One of the best prospective randomized trials of combination chemotherapy for advanced disease came from a US intergroup study in which ifosfamide was or was not given to previously untreated patients with metastatic or advanced STS receiving doxorubicin plus dacarbazine. This study demonstrated no survival advantage for the group receiving ifosfamide in combination with doxorubicin plus dacarbazine, although this group had a statistically significant increase in objective response rate.178, 179 The role of combination chemotherapy is further called into question for broad use given the statistically significant increase in toxicities when ifosfamide was added. Thus, despite the increased anticancer activity as evidenced by the small but significant improvement in response rates, no survival benefit was obtained by adding a third drug.
In a similar manner, the addition of higher dose ifosfamide (10 g/m2 per cycle) to doxorubicin (75 mg/m2 per cycle) was associated with a statistically significantly higher RECIST response rate (26% vs 14%), median progression-free survival rate (7.4 months vs 4.6 months), greater toxicity burden, and a trend to a survival advantage (14.3 months vs 12.8 months, p = 0.076).180 It was not clear from these data if sequential use of the two agents would yield similar outcomes to combination treatment. The survival data in particular serve as a touchstone by which we determine success for therapy in metastatic sarcomas. The data from these two randomized trials support the use of combination chemotherapy for patients with symptomatic disease who are in need of a response; by the same token, it is not unreasonable to use single agents sequentially to minimize patient toxicity for less symptomatic or asymptomatic patients who remain in need of therapy.
Strategies to improve the therapeutic index of chemotherapy
Dose intensification using stem cells
An obvious strategy to increase response rates has been to increase dose intensity, adding stem cell support, as examined in hematological malignancies, breast adenocarcinoma, and other malignancies. This was initially attempted solely with provision of autologous bone marrow support and in the past decade has been significantly facilitated by the availability of hematopoietic cytokines to improve hematologic tolerance to myelosuppressive chemotherapy. It is clear that peripheral blood progenitor cells can be mobilized and harvested following standard chemotherapy for sarcoma supported by granulocyte colony-stimulating factor.181 Testing the limits of high-dose therapy, full-dose doxorubicin, ifosfamide, and cisplatin are possible with stem cell support not clearly better than standard dosing in osteogenic sarcoma.182 Even in chemotherapy-sensitive diseases such as Ewing sarcoma, dose intensification with autologous stem cell support does not appear to be beneficial for improving survival, although contamination of stem cells with tumor cells may be a contributing factor to its lack of efficacy. The use of high-dose chemotherapy with stem cell and cytokine support remains investigational for sarcomas.
Encapsulated anthracyclines
Another strategy to increase the therapeutic index of anthracyclines is to encapsulate the drug within a liposomal vehicle. At least three liposomal preparations of anthracyclines have been tested, and all have shown some efficacy against sarcomas. Notably, the most widely used agent has decreased cardiac risk in comparison to older preparations of larger liposomes containing doxorubicin. Pegylated liposomal doxorubicin (Doxil/Caelyx) is a small liposome with polyethylene glycol anchored within the lipid bilayer, acting as a hydrophilic coating to preserve the circulating half-life of the liposome and prevent degradation within the reticuloendothelial system. This preparation, given at a dose less than that of unencapsulated doxorubicin, is better tolerated than doxorubicin, with substantially less myelotoxicity, cardiac toxicity, and alopecia at the cost of hand–foot syndrome and idiosyncratic reactions to the first dose of therapy; while its dose is lower, it has long half-life in the circulation (30–70 h), leading to an area under the curve for 50 mg/m2 that is 300 times that of doxorubicin itself. In a randomized phase 2 study, pegylated liposomal doxorubicin demonstrated similar activity to normal doxorubicin (9% vs 10% response rate in the era before GIST was recognized as a separate type of sarcoma); formal noninferiority or equivalence was not determined, but pegylated liposomal doxorubicin was significantly less toxic than doxorubicin.183 Less toxic anthracycline preparation has yielded a way to extend systemic therapy to patients with poorer performance status and in principle is a novel way of treating very slowly growing connective tissue lesions such as myxoid liposarcomas or desmoid tumors.
Beyond anthracyclines and ifosfamide
Few other agents beyond doxorubicin and ifosfamide demonstrate activity in soft tissue sarcoma, but this is a generalization in an era in which more histology specific data are needed for treatment decision making. It is recognized that dacarbazine (DTIC) has minor activity in STS, and studies have demonstrated that leiomyosarcoma and to a lesser degree solitary fibrous tumor are two diagnoses in which dacarbazine and its orally absorbed relative temozolomide have greatest activity.184–186 Gemcitabine and docetaxel as a combination was first demonstrated to have activity in uterine leiomyosarcoma, after relatively disappointing phase 2 trials of gemcitabine as a single agent, suggesting true synergy between the two agents.187, 188 Randomized study of gemcitabine and docetaxel showed the combination was superior to gemcitabine alone with respect to both PFS and OS in a group of unselected sarcomas.189 Notably, responses were most common in UPS and pleomorphic liposarcoma, more than leiomyosarcoma. A randomized study demonstrated the superiority in PFS and OS of gemcitabine and dacarbazine over dacarbazine alone, the second in which gemcitabine combinations were superior to single agents in important radiological and clinical outcomes.190 The addition of bevacizumab to the gemcitabine–docetaxel combination did not add to the chemotherapy backbone, and progression-free survival was numerically, although not statistically, inferior to treatment with the two chemotherapy drugs alone.191 Thus, either the gemcitabine–docetaxel or gemcitabine–dacarbazine combinations are good options after failure of doxorubicin and/or ifosfamide. Indeed, the perception of toxicity of gemcitabine-based therapy versus doxorubicin-based therapy has led to its acceptance in first line in metastatic sarcoma patients in the United States.192 The author generally employs gemcitabine–dacarbazine for leiomyosarcomas and gemcitabine–docetaxel in UPS, pleomorphic liposarcoma, and pleomorphic liposarcoma, given the responses seen to date on and off trial.
Newer agents
Sarcomas represent a fertile ground for the field of drug development. Doxorubicin was first recognized as an effective agent against sarcomas and subsequently was developed into one of the most widely used anticancer agents ever discovered. Imatinib and its spectacular results in a chemotherapy-resistant diagnosis provided a proof of principle that has since been borne out in other solid tumors with EGFR inhibitors, BRAF inhibitors, and the like. A few of newer agents and their utility in sarcoma are noted below. The next edition of this chapter will no doubt contain much more information on new drug and new targets in a specific histology or molecularly characterized subset of sarcomas, much as is happening across medical oncology.
Trabectedin (ET-743, ecteinascidin)
Trabectedin is a marine-derived drug from the marine tunicate Ecteinascidia turbinata. It covalently binds to the minor groove of the DNA and blocks the cell cycle in late S and G2 and affects the transcription in part by prevention of binding of transcription factor NF-Y, thus decreasing expression of a variety of genes, including multidrug resistance genes. After initial promising results in phase 1 studies, multiple phase 2 trials were performed, the most successful of which (a randomized phase II study of two schedules of drug) led to the drug’s approval in Europe for refractory STS.193, 194 A follow-up randomized study showed greater activity of trabectedin than dacarbazine.195
The randomized phase 2 study of trabectedin in patients with metastatic leiomyosarcoma and liposarcoma compared a weekly schedule of drug with a 24-h infusion schedule.193 In this 270 patient study, the longer 3-weekly infusion was associated with median time to progression of 3.7 months versus 2.3 months, p = 0.03; OS showed a numerically longer median OS for the 3-weekly infusion, 13.9 months versus 11.8 months, p = 0.19. Particular activity is noted in myxoid–round-cell liposarcoma, although activity in other histologies has been observed.
In a similar manner, the microtubule-targeted agent eribulin demonstrated activity in a phase 2 study,196 which led to the conduct of a study similar to that of the trabectedin trial above, in which eribulin was tested against dacarbazine in a phase 3 trial in leiomyosarcoma and liposarcoma. The phase 3 data confirmed the activity seen in phase 2. The OS was not significantly different between trabectedin and dacarbazine in this study (12.4 months vs 12.9 months), there was a significant improvement in PFS with trabectedin over dacarbazine (median 4.2 months vs 1.5 months) in this randomized phase III trial.
Beyond cytotoxics, the most important result to date involves the SMOKI pazopanib. After the demonstration of activity in a phase 2 trial,197 a phase 3 study termed PALETTE was conducted, involving 369 patients with STS progressing after standard therapy. With a 2 : 1 randomization for pazopanib 800 mg versus placebo, the pazopanib arm demonstrated statistically superior PFS. Median PFS in the placebo arm was 4.6 months in the pazopanib arm and 4.6 months in the pazopanib arm, with a corresponding HR of 0.35 (p < 0.001) as assessed by independent radiology review. Median OS at final analysis was 10.7 months in the placebo arm versus 12.6 months in the pazopanib arm, HR = 0.87, p = 0.26.198 These data were sufficient to obtain broad international approval for pazopanib for metastatic STS worse after standard therapy.
Increasingly, the molecular characteristics of specific sarcomas will dictate therapeutic options. On the basis of the tumor DNA alterations (gene loss, translocation, or mutation), mTOR inhibitors such as sirolimus have activity in perivascular epithelial cell tumors (PEComas),199 and imatinib and other inhibitors of PDGF receptor and CSF1 receptor have activity in DFSPs200 and tenosynovial giant cell tumor (TGCT).201, 202 Crizotinib, the ALK inhibitor, is active in ALK translocation positive inflammatory myofibroblastic tumor (IMT). There will be increasing numbers of such agents demonstrating activity as targets other than kinases come into focus therapeutically, perhaps including chondrosarcomas with their frequent IDH1 and IDH2 mutations, and synovial sarcoma, Ewing sarcoma, and MPNSTs, which appear to have alterations in epigenetic regulators PRC1, LSDs, and BRD4, respectively, each becoming targetable imminently as are other epigenetic regulators such as EZH2.
Management of local recurrence
If an isolated LR is identified, the treatment goals are the same as for patients with primary tumors, namely, optimal local control while maintaining as much function and cosmesis as possible.107 Early identification of local relapse may improve the chance of successful salvage therapy, and, like newly diagnosed patients, these patients are probably best managed in specialized multidisciplinary sarcoma centers. An approach to the evaluation and management of locally recurrent STS is summarized in Figure 14. The initial evaluation must include a full review of previous therapy because this will have a bearing on the therapeutic options available. Therefore, all prior surgery and pathology reports should be examined, as should reports on previous chemotherapy and previous RT, especially volume treated, dose, and energy of radiation.
Figure 14 Schema for approaching the patient with local recurrence of STS. The schema is oriented toward extremity lesions but is equally applicable to other anatomic sites (e.g., head and neck and retroperitoneum). Abbreviations: BRT, brachytherapy; EBRT, external beam radiotherapy.
Source: Catton et al. 1999.107 Reproduced with permission of Elsevier.
Staging should be performed in the same way as for newly presenting patients. The areas adjacent to the original lesion and potentially contaminated by previous surgical interventions should be scrutinized carefully. Both these areas and tissues adjacent to the recurrent tumor, containing potential tumor extensions, should be considered at risk and candidates for resection and/or inclusion in radiation fields.
Several distinct groupings are evident under the rubric of “locally recurrent” disease: (1) cases in which prior treatment did not include RT, (2) cases treated with RT in the past, (3) cases in which distant metastases are also present, and (4) cases in which it is difficult to distinguish between recurrence and secondary tumors induced by RT. Although the therapeutic options available are more limited in recurrent disease and the challenge posed by these cases are much more formidable, a proportion of these patients can be cured. Clinical experience is needed to determine which therapeutic options are appropriate in a given case of recurrent disease.
Gastrointestinal stromal tumors
No discussion of sarcomas would be complete without noting the remarkable advances made with one particular sarcoma subtype, GIST, which has changed the way people think about solid tumors. With the recognition of KIT as a good marker for GIST to distinguish it from other sarcomas and the recognition of KIT or PDGFRA activating mutations that are likely responsible for the constitutive activation of KIT,203 clinical studies followed rapidly and have been done in parallel to studies investigating the biology of GIST. Of note, presence of an immunohistochemical marker cannot be equated with tumor response, as is suggested by companies that purport this to be the case. Ewing sarcomas can mark positive for KIT by immunohistochemistry, but KIT is not mutated in these tumors and Ewing sarcomas do not respond to imatinib.
Surgical principles for management of primary GIST differ from those for other sarcomas and visceral adenocarcinomas. In general, resection requires a minimal margin of normal tissue, not the wide margins of other sarcomas or visceral adenocarcinomas. Unlike gastric cancer, GIST do not generally metastasize to local–regional lymph nodes (except in some GIST arising in the pediatric population), rendering lymph node dissection unnecessary in the vast majority of patient. Thus, gastric GISTs can often be removed by simple wedge resections, and large gastric resections are only rarely required, generally due to anatomic constraints. Similarly, GISTs arising in the small intestine, the colon, or the rectum may be resected with minimal margins. Furthermore, patients with GIST undergoing a macroscopically complete resection and with positive microscopic margins are not at increased risk for LR compared to those resected with negative margins.204
After the recognition of in vivo efficacy of imatinib in a GIST cell line,203 treatment of metastatic disease has rapidly advanced from treating a single patient to phase 1, 2, and 3 studies for patients with metastatic disease.205–209 The results have been remarkably consistent. Imatinib is at least 10-fold more active than any agent ever examined for treatment of GIST (formerly called GI leiomyosarcoma). The response rate to imatinib is approximately 50%, 30–35% with stable disease, and ∼15% with overt progression on therapy. The US phase 3 data indicate that 400 and 800 mg yield equivalent time-to-progression curves, but the European/Australian phase 3 study indicates that time to progression is improved with the higher dose (800 mg daily) arm.206 Remarkably, patient kinase genotype determined relative sensitivity to therapy (Figure 15).210 Patients with KIT exon 11 mutation had an 80–90% response rate, while patients with KIT exon 9 mutation had a high response rate only one-third to one-half. Patients with no mutation in KIT or PDGFRA had a much lower response rate, but still higher than that observed for any other chemotherapy drugs. For the time being, regardless of mutation status, imatinib remains the first-line standard of care for metastatic GIST. A dosage of 400 mg daily is a reasonable starting point for most patients, with increase toward 800 mg if there is evidence of progression of disease or the presence of exon 9 mutation.211 Therapy should not be interrupted if new hypodense lesions appear in the liver; these likely represent occult metastatic disease, as has been borne out with radiological and clinical experience.176
Figure 15 Mutation status of gastrointestinal stromal tumors and location on the KIT or PDGFRA protein.
The median time to progression for patients with metastatic GIST on imatinib is approximately 2 years. Patients with progression on a lower dose of imatinib can respond further with dose increases. The phase 1 study of imatinib indicated that the maximum tolerated dosage is 800 mg daily (400 mg by mouth bid). Patients with progression of disease have had unusual patterns of progression, the so-called tumor within a tumor, which represents clone(s) with second resistant KIT mutations.160 In some cases, tumor regrowth in an apparently necrotic tumor is observed, and in others only one metastatic deposit is seen to progress instead of the multiplicity of lesions seen in many patients with advanced GIST. As a result some patients have been treated with carefully planned operations to remove problematic individual sites of metastatic disease.
The rationale for consideration of metastasis surgery for patients with responding or stable metastatic disease on imatinib is based on the observations that (1) pathologic complete response to imatinib is very rare (<5%) and many (perhaps most) patients will eventually develop secondary resistance to imatinib owing chiefly to the development of secondary resistance mutations. Reports from high-volume centers demonstrate that carefully selected patients treated by imatinib and subsequent metastasectomy appear to have very favorable progression-free survival rates. Whether this is due to case selection or bona fide clinical benefit related to surgery is unclear.212, 213 Randomized controlled trials of the impact of metastasectomy in patients with stable or responding disease were planned in the United States, Europe, and China. Poor accrual led to the closure of the European and Chinese trials, and the American trial was never started.
The utility of imatinib in metastatic GIST spawned new SMOKIs, some of which have been through phase 3 trials with subsequent regulatory approval. In the setting of imatinib resistance for metastatic disease, the addition of mTOR inhibitor everolimus to imatinib demonstrated minor activity.214 However, greater activity has been observed with other KIT-targeted SMOKIs. Sunitinib is active in imatinib-resistant GIST and is associated with both significantly improved progression-free survival and OS in comparison to placebo.215 With the demonstration of activity of sorafenib in GIST,216, 217 regorafenib (a fluorinated form of sorafenib) was found active in phase 3 trial of drug against placebo, in which better attention was paid to the placebo group; in this so-called GRID study, the patients on placebo were crossed over more rapidly to drug than the study involving sunitinib, perhaps accounting for the lack of OS benefit seen in the GRID study.218 After failure of other agents, some form of SMOKI appears to be indicated for further therapy; use of imatinib was superior to placebo in late stage patients in a randomized trial.219
Given the remarkable activity of imatinib in the metastatic setting, it is not surprising that imatinib has been tested in the adjuvant setting as well. Data for studies of 0 versus 1 year of imatinib, 0 versus 2 years of imatinib, and 1 year versus 3 years of imatinib have all consistently demonstrated the benefit of the longer course of therapy. Adjuvant imatinib was initially approved in the United States by virtue of the ACOSOG Z9001 study, in which 1 year of imatinib (400 mg/daily) was compared to placebo following complete resection of GISTs >3 cm in size. This study was halted to further accrual after accruing 762 patients when a planned interim analysis for the primary endpoint demonstrated superior progression-free survival in the imatinib arm (97% vs 83% in the placebo arm).220 A randomized trial comparing 1 year versus 3 years of adjuvant imatinib subsequently demonstrated improvement not only in the recurrence-free survival but also in the OS, favoring longer duration of adjuvant therapy.221 Whether longer adjuvant treatment with imatinib for longer periods adds further benefit is uncertain and is the subject of a single-arm 5-year trial of imatinib. At this juncture, the best recommendation that can be made is that 3 years of adjuvant treatment be considered for patients with intermediate- and high-risk resected primary GISTs. Risk is well stratified by clinical, pathological, and molecular features of the primary tumor.222–224
Locally aggressive lesions: DFSPs, tenosynovial giant cell tumor (TGCT), perivascular epithelial cell tumor (PEComa) as targets for small molecule oral kinase inhibitors
DFSP is a nodular “protuberant” lesion arising from the dermis with characteristically slow but persistent growth over many years. Although histologically of low grade or borderline malignant potential, DFSP has a propensity for LR after simple excision. A chromosomal translocation t(17;22) and gene fusion product (Table 1) result in the expression of a COL1A1-PDGFB fusion protein that is processed to mature PDGF-B, resulting in apparent autocrine or paracrine interaction with the PDGF-B receptor on the cell surface of DFSP. Notably, imatinib inhibits the PDGF tyrosine kinase receptor in a similar manner that it inhibits the BCR-ABL tyrosine kinase receptor of chronic myeloid leukemia and KIT kinase of GIST. In recurrent DFSP, imatinib has activity, confirming that kinases can be very good targets in solid tumors, even rare ones, if the genetics of the tumor are suggestive.200, 225–227
For a similar reason, the t(1;2) COL6A3-CSF1 generates a fusion protein that is responsible for the aggressive inflammatory infiltrate of TGCT (formerly termed pigmented villonodular synovitis). Imatinib has activity against CSF1R (FMS, CD115) and can cause recurrent lesions to shrink.201 More specific SMOKIs or monoclonal antibodies against CD115 can also shrink recurrent TGCT lesions. Finally, PEComa, found in the genetic syndrome tuberous sclerosis, and the related lesion lymphangioleiomyomatosis lack either TSC1 or TSC2, tumor suppressor genes. TOR (target of rapamycin) is activated downstream after TSC1 and TSC2 inactivation, and thus it is not surprising that SMOKI sirolimus and structurally related compounds are active in these conditions, as further proof of principle of blockade of a “driver” kinase in a solid tumor can yield radiological and clinical benefit for patients.199
Retroperitoneal sarcomas
RPS comprise about 15% of STS. RPS present late and are located in regions where the administration of both surgery and RT is often compromised (e.g., adjacent to the small bowel and the liver). Consequently, the local control rates achieved with combined-modality treatment of extremity STSs are not seen in RPS.54, 228–232 For example, in a series of 102 RPS patients treated at Princess Margaret Hospital, complete excision was achieved in only 45, gross disease remained in 29, and only a biopsy was possible in 28.232 The overall local–regional relapse-free rates were 28% and 9% at 5 and 10 years, respectively. RT did not improve survival but appeared to significantly lengthen the time to local–regional relapse, especially with higher doses. Complete tumor resection was the only significant prognostic variable for survival and local–regional and distant failure, similar to other series referenced above. RPS patients should be evaluated in a multidisciplinary clinic before treatment so that patients can benefit from the expertise and investigational approaches available in such centers.
Controversial reports from Europe explored extended surgery termed compartmental resection as a strategy to improve local control for patients with RPS.233, 234 These retrospective reports from France and Italy have suggested that local control may be enhanced by resecting adjacent involved viscera—primarily the kidney and the colon. Interpretation of these reports is complicated by significant selection bias and, as outlined in an editorial that accompanied these papers,235 no specific therapeutic recommendation could be made based on these data. At this time, there are no clinical trials demonstrating improved local control with more radical surgery that involves resection of adjacent uninvolved viscera.
Pre- and postoperative radiotherapy approaches
A variety of adjuvant RT approaches have been used for RPS. One approach, evaluated in a prospective randomized trial, used an IORT boost (20 Gy) to the tumor bed followed by postoperative external beam (35–40 Gy); this approach was compared with conventional postoperative RT (50–55 Gy). In this study of 35 patients, the incidence of local–regional recurrence was lower in the experimental treatment arm, but no improvement in survival was demonstrated.236 IORT was associated with a high rate of peripheral neuropathy when large, sometimes overlapping, RT portals were used to cover the sacral plexus region. However, gastrointestinal complications were more common in the control group, in whom higher doses were delivered to the bowel.
Other researchers have investigated strategies employing preoperative RT. Gieschen et al.237 from the Massachusetts General Hospital reported on 37 patients who underwent preoperative RT, resection, and then, when feasible, electron beam IORT. The grossly complete resection rate was 83%, and the 5-year actuarial OS, DFS, local control, and freedom from distant disease rates were 50%, 38%, 59%, and 54%, respectively. Earlier reports from the same group described a 70% complete resection rate and 81% 4-year local control rate. The more recent report also indicates that complete resection and IORT improved OS and local control rates (74% and 83%, respectively) compared with no IORT (30% and 61%, respectively). However, the study was not randomized, and potentially more favorable and accessible lesions may have been chosen for IORT. Petersen and colleagues at the Mayo Clinic found improved local control, at least of primary tumors, using a similar treatment approach.238
The University of Toronto Sarcoma Group described an unusually favorable outcome, especially for primary lesions, in a single-arm prospective trial of preoperative RT (median dose of 45 Gy in 25 fractions) plus postoperative BRT in selected cases.239 Of interest, acute toxicity resulting from preoperative RT was differentiated prospectively from the effects of other treatments. Although the median radiation volume exceeded 7 L, preoperative external beam RT was associated with extremely low gastrointestinal toxicity scores. Furthermore, no patient was hospitalized for acute toxicity, and there were no treatment interruptions or cessations of treatment because of acute toxicity. The remarkably low toxicity of the preoperative RT with enormous volumes has been attributed to the displacement of the bowel outside the target volume. In contrast, the selective use of postoperative BRT was associated with toxicity, and there was little evidence that BRT contributed to the enhanced tumor outcome reported.
The University of Texas MD Anderson Cancer Center and Princess Margaret Hospital groups combined the data from their phase 2 and pilot studies. This pooled analysis demonstrated very favorable OS rates for patients treated with preoperative radiation combined with surgical resection.240 However, we again caution against overinterpretation of results that could be explained by surgical technique at major referral centers or by case selection. Similarly, it remains unclear what contribution was provided by the BRT or IORT, which should probably remain protocol based or be reserved for nonstandard use in selected cases. Notably, however, the preoperative RT approach seems to continue to demonstrate favorable local control and OS in very long-term follow-up of the PMH study.
The results of these three reports add credence to the advantages posited for preoperative RT in RPS: (1) the tumor bulk often displaces the small bowel from the high-dose RT region, resulting in a safer and less toxic treatment; (2) the bowel is unlikely to be fixed by surgical adhesions as when RT is given postoperatively, enabling safe delivery of a higher dose to the true area at risk; (3) optimum knowledge of the gross tumor location is possible, permitting better radiation targeting; (4) the tumor is contained by an intact peritoneal covering, providing a physical barrier to immediate tumor dissemination; (5) the risk of intraperitoneal tumor dissemination at the time of surgery may be reduced by the biologic impact of preoperative RT; and (6) using traditional principles of sarcoma RT, the radiation dose believed to be biologically effective is lower in the preoperative setting. Although RT planning for RPS can be complex, the use of conformal techniques or intensity modulation usually permits the RT dosage to be administered safely to the critical organ preoperatively. It is imperative that members of both the surgical and radiation oncology teams be present in the operating room when a significant amount of the liver has been irradiated preoperatively to evaluate the planned residual liver volume. We cannot overemphasize the detailed evaluation of dosimetry and treatment planning films are needed to be certain that a sufficient volume of unirradiated liver is left unresected at the time of surgery.241
The only standard that exists at present for RPS treatment is that complete surgery should be performed wherever possible. The adjuvant RT approaches described above should be considered experimental and be the subject of further assessment. Recently, it was also suggested that a more scientifically sound approach to study design should be undertaken in RPS, particularly exploring the role of external beam RT through a randomized controlled trial.174 The European Organisation for Research and Treatment of Cancer (EORTC) is currently conducting a phase 3 randomized study of preoperative RT plus surgery versus surgery alone (NCT01344018) that is readily on target for completion.242
Chemoradiation approaches
Although chemotherapy alone has not been associated with obvious improvements in outcome of RPS, chemoradiation, as in other solid tumors, is a subject of interest as a treatment strategy for RPS. This is especially relevant in high-grade RPS, which have a more adverse prognosis than the more common low-grade lesions.
Pilot studies using concurrent preoperative chemotherapy and external beam radiation have been reported, often in patients with extremely large tumors, with acceptable toxicity and with achievement of local control in patients in whom a negative-margin resection was possible.141, 243 These reports demonstrate that chemoradiation approaches are feasible. Additional phase 2 studies will be necessary to clarify response rates and toxicity profiles and determine whether chemoradiation should be tested in phase 3 trials. As discussed earlier, the Radiation Therapy Oncology Group completed a phase 2 study of preoperative doxorubicin and ifosfamide followed by preoperative RT and then by surgical resection with an intra- or postoperative radiation boost in patients with intermediate- and high-grade RPS. This study demonstrated significant toxicities and event-free outcomes that were considered modest.144, 146
Additional issues in STS management
Functional outcome and morbidity of treatment
The functional result of extremity sarcoma management has become an important component of outcome assessment. Assessing the functional result is difficult, as it requires methods that are valid and reproducible. Many centers have yet to become experienced in the development and use of these methods, and much of the literature contains significant heterogeneity in patient samples.
Thus far, the variables associated with poorer functional outcome include large tumor size, higher doses and larger volumes of radiation, nerve sacrifice, postoperative fracture, and wound-healing complications.244, 245 To evaluate and compare functional outcome, it is imperative that functional data be reported consistently. Three disease-specific scoring scales have been reported as useful in assessing functional outcome.246 This area has been discussed in detail by Davis, who observed that “function” has many meanings in the literature.246 The concepts of impairment, disability, and handicap following extremity STS are likely misunderstood and certainly not used consistently. Davis noted that impairment is a disorder of structure or function whereas disability is a restriction or lack of ability to perform an activity. Handicap results from impairment and disability and prevents or restricts an individual from performing in a role that is normal for the individual. For sarcoma patients, impairments can be manifested as soft tissue fibrosis, loss of motion at a joint, and decreased muscle strength; disability can be manifested as limited mobility and difficulty performing routine self-care and activities of daily living; and handicap can be evident in limitation in family roles, social functioning, and the capacity for employment.
Impairments are the most frequently reported deficits following limb-preserving therapy for extremity STS, and up to 50% of patients appear to experience significant impairments.246 Disability occurs less frequently, although reports are contradictory. It seems likely that many sarcoma patients learn to accommodate their impairments. Handicap has received little attention in the literature. However, the limited data suggest that up to 50% of patients may experience changes in their employment and vocation status after treatment of extremity STS. The continuing challenge in treating sarcomas is to define the therapeutic ratio for the patient with sarcoma of the extremity. Specifically, the aim of the multidisciplinary team will be to minimize the amount of treatment while maintaining or improving current standards of disease control to reduce treatment morbidity and enhance patient outcome.
Wound complications
Considerable variability in reporting wound-healing complications exists in the literature. WCs have been reported in up to 40% of patients undergoing extremity sarcoma surgery.85, 247–249 Differences in the definition of WCs probably account for some of the variability in reporting. The retrospective data suggest that factors associated with compromised wound healing include advanced patient age, poor nutritional status, lower extremity tumor location, large tumor size, and preoperative adjuvant treatment, especially RT.247, 248, 250 Particularly high complication rates were noted with combination preoperative RT and hyperthermia.117 Although many authors have reported an association of WCs with preoperative RT, reports of high rates of surgical complications without RT or chemotherapy also exist.251 Most likely, these relate to the risk of major WCs associated with extensive tumor resection, particularly in the lower extremities. The use of vascularized tissue transfers to replace resected tissues and optimize wound closure may decrease the risks of major WCs and allow for more extensive limb-sparing surgical approaches.250, 252, 253 As noted earlier, the SR2 trial results have confirmed the adverse effect of preoperative RT on wound healing in a prospective manner, but did not resolve the contribution of tissue transfer for wound reconstruction as its use was determined on an individual basis at the surgeon’s discretion.96
Molecularly and pathobiologically based sarcoma management
Management of sarcomas is increasingly being driven by the specific nature of the disease entity, most importantly the pathophysiologic subtype. The work of Pasteur and Koch was fundamental for the recognition and definition of pathogenic microbes; similarly, many laboratories today are identifying molecular lesions that will redefine the field of sarcoma research. An example of this work is in the recognition of soft tissue Ewing sarcoma/PNET family of tumors as relatives of their bony primary counterparts by virtue of the same spectrum of translocations in each clinical setting. These tumors should be treated with curative intent using an aggressive multimodality approach that begins with multiagent chemotherapy. If primary surgery has removed measurable disease, adjuvant chemotherapy is definitely indicated, with consideration of adjuvant RT. By adopting similar strategies for PNETs and Askin tumors as for conventional Ewing sarcomas, outcomes improved. The molecular similarities between tumors of this family have led to the current convention of considering them morphologic and clinicopathologic variants of the same underlying molecular disease process.254, 255
Greater understanding of the biology of translocation gene products points to epigenetic events being important in sarcomagenesis and should impact on the efficacy of chemotherapy. Ewing sarcoma and synovial sarcoma are chemotherapy-responsive STS subtypes. While TP53 could be posited as a reason that certain sarcomas are chemotherapy sensitive, TP53 status is not the only factor dictating chemotherapy sensitivity as there are other TP53 wild-type sarcomas that are far less sensitive to chemotherapy, for example, extraskeletal myxoid chondrosarcoma and alveolar soft part sarcoma. Striking research on the epigenetic mSWI/SNF (BAF) complexes and their role in synovial sarcoma hopefully will shed light on some of the basic mechanistic aspects permitting synovial sarcoma survival, in order to better engender tumor cell death.256, 257
As a final example, the multiple histopathologic subtypes of liposarcomas are becoming increasingly well researched and well understood, and mechanisms by which some forms acquire their characteristic DNA signature are beginning to become understood.258 Myxoid and round-cell liposarcomas usually exhibit a characteristic chromosomal rearrangement t(12;16) (q13;p11) FUS-DDIT3. These liposarcomas tend to be relatively sensitive to chemotherapy, with trabectedin standing out as most active in this specific diagnosis. Trabectedin appears to inhibit the binding of the FUS-DDIT3 fusion protein to DNA, which then causes liposarcoma cell death or differentiation.259, 260 The rarer pleomorphic liposarcoma subtype has more in common with undifferentiated pleomorphic sarcoma than other liposarcomas. Finally, well-differentiated liposarcomas exhibit ring and giant marker chromosomes on cytogenetic analysis and these karyotypic abnormalities, often involving massive amplification of chromosome 12q, carry through in dedifferentiated liposarcomas. Unraveling the ability of the CDK4 and HDM2 loci on 12q to amplify hundreds of times in well-differentiated–dedifferentiated liposarcoma may provide insight on how better to attack these relatively chemotherapy-insensitive sarcomas.261–263
Immunotherapy for STS
As of the time of this publication, the examination of the immune system as treatment for sarcoma cannot even be considered in its infancy, but rather in a prenatal state. Immunotherapy is already approved in some countries for osteogenic sarcoma, with the nonspecific immune adjuvant mifamurtide (MTP-PE) improving the cure rate when used as part of adjuvant treatment.120 While the proof of principle of engineered T-cell therapy in NY-ESO-1-positive sarcomas such as synovial sarcoma already has been demonstrated by Robbins et al.,264, 265 the potential benefit of immune checkpoint blockade is still very much unclear, as no studies were able to be started in sarcomas until 2015. If the hypothesis holds that the cancers with the most DNA mutations and alterations will be most responsive to immunotherapy, then we expect to see undifferentiated pleomorphic sarcoma, leiomyosarcoma, and osteosarcoma among the best responders and much less activity in translocation-associated sarcomas. Conversely, CAR T-cell therapy may be more easily targeted to translocation-associated sarcomas if an appropriate target antigen can be identified.
Summary
It is clear that STS management has improved in the past 10 years. In fewer than three decades, the standard of care has shifted toward coordinated multimodality care in specialty centers, with increased rates of function-sparing surgery and better outcomes for patients. Judicious use of aggressive multimodality approaches shows promise to decrease relapse rates and improve survival rates. The genomic and immunological revolutions in cancer are furthering the fundamental understanding of these unusual diseases and providing novel approaches for diagnostic techniques, which will banish the vagaries and lack of consistency that have plagued this field of clinical investigation and also will provide resources for selecting new targets for therapy. New therapeutic initiatives are attacking the basic mechanisms of sarcomatous transformation of cells in some subtypes of STS, and it is hoped that these initiatives will improve outcomes for patients with less morbidity than current treatments entail. Large collaborative studies should further this work.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


