INCIDENCE
Sarcomas are a rare and heterogeneous group of tumors that arise from mesenchymal tissues. According to the estimates of the American Cancer Society, approximately 1.6 million people were estimated to be diagnosed with cancer in the United States in the year 2013, with only 14,420, or just less than 1% of cases, representing sarcomas (1); 11,410 of these cases represent new soft tissue sarcomas and 3,010 represent bone sarcomas, with 4,390 and 1,440 deaths, respectively, resulting from these tumors (1).
EPIDEMIOLOGY AND PATHOGENESIS
The etiology and pathogenesis of sarcomas is not well understood. Multiple environmental factors including radiation and chemical exposures, trauma, and infection have been associated with the development of soft tissue and bone sarcomas. Both sporadic and inherited molecular and genetic aberrations have been identified in specific subsets of sarcoma and have been implicated in sarcomagenesis.
Several inherited familial cancer syndromes have been associated with a predisposition to development of soft tissue and bone sarcomas. The Li-Fraumeni syndrome resulting from a germline mutation of the p53 tumor suppressor gene is associated with increased risk of soft tissue sarcomas and osteosarcoma among several other cancers in children and young adults (2). Inherited retinoblastoma is also associated with the development of sarcomas, both osteosarcoma and soft tissue sarcoma (2). Neurofibromatosis type 1 is associated with an increased risk of development of sarcoma in a preexisting neurofibroma, resulting in a malignant peripheral nerve sheath tumor (3). Sarcomas have been associated with other cancer family syndromes including basal cell nevus syndrome, Werner syndrome, familial adenomatous polyposis, and Gardner syndrome, among others (2).
Recurrent cytogenetic abnormalities have been identified in many sarcomas and are thought to contribute to sarcomagenesis. Genetic profiling of bone and soft tissue sarcomas has expanded the characterization of mesenchymal tumors beyond translocation-positive/translocation-negative categorizations to identify three distinct groups: (1) genetically unstable sarcomas with complex karyotypes; (2) tumors with specific, recurrent genetic alterations such as translocations, deletions, or copy number variations; or (3) tumors with molecular aberrations such as amplifications, mutations, or loss of heterozygosity (Table 42-1). These alterations serve an important role in diagnosis as well as prognosis and have potential implications for therapy.
| Tumor | Cytogenetic Abnormality | Gene Product |
|---|---|---|
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) t(1;13)(p36;q14) t(X;2)(q13;q35) | PAX3-FOXO1A PAX7-FOXO1A PAX3-AFX |
| Alveolar soft tissue sarcoma | t(X;17)(p11.2;q25) | ASPL-TFE3 |
| Clear cell sarcoma | t(12;22)(q13;q12) t(2;22)(q32;q12) | EWSR1-ATF1 EWSR1-CREB1 |
| Congenital fibrosarcoma | t(12;15)(p13;q25) | ETV6-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1-PDGFB |
Desmoplastic small round cell tumor Epithelioid hemangioendothelioma | t(11;22)(p13;q12) t(1;3)(p36;q25) | EWSR1-WT1 WWTR1-CAMTA1 |
| Endometrial stromal sarcoma | t(7;17)(p15;q11) | JAZF1-SUZ12 |
| Gastrointestinal stromal tumor | Activating mutations Overexpression Loss of 14q LOH of 22q Loss of 1p | KIT, PDGFR, BRAF ETV1 |
| Inflammatory myofibroblastic tumor | t(1;2)(q22-23;p23) t(2;19)(p23;p13.1) | TPM3-ALK TPM4-ALK |
Low-grade fibromyxoid sarcoma
Lipoma | t(7;16)(q32–34;p11) t(11;16)(p11;p11) t(3;12)(q27-28, q14-15) | FUS-CREB3L2 FUS-CREB3L1 HMGA2-LPP |
| Malignant peripheral nerve sheath tumor | Inactivating | Deletion of NF1 |
| Myxoid chondrosarcoma | t(9;22)(q22-31;q11-12) t(9;17)(q22;q11) t(9;15)(q22;q21) | EWSR1-NR4A3 TAF15-NR4A3 TCF12-NR4A3 |
| Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-DDIT3 |
| t(12;22)(q13;q12) | EWSR1-DDIT3 PIK3CA mutation | |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18-SSX1 SS18-SSX2 SS18-SSX4 |
| WDLPS/ALT and DDLPS | 12q14-15 (supernumerary ring chromosomes; giant marker chromosomes) | Amplification of MDM2, CDK4, HMGA2, SAS, GLI, and JUN |
| Uterine leiomyosarcoma | t(12;14)(q7-) |
Molecular testing has become more widely used for diagnosis in soft tissue sarcoma, identifying specific abnormalities in certain histologic subtypes (4). For example the t(x;18) translocation is a specific marker for synovial sarcomas. The transcript formed from this translocation (SYT-SSX) can be detected using polymerase chain reaction (PCR) testing. In one study, the SYT-SSX transcript was positive in 84.5% of tumors where the diagnosis of synovial sarcoma was certain based on histology and 24.3% of tumors in which synovial sarcoma was considered but not first in the differential diagnosis (5). Another example is the FUS-DDIT3 translocation in myxoid liposarcoma where the presence of DDIT3 rearrangement confirms the diagnosis (6). These data suggest that molecular pathology may also be a useful adjunct in the diagnosis of some sarcomas.
Specific translocations have also been investigated as prognostic biomarkers and potential therapeutic targets. For example, the EWS-FLI1 transcript in Ewing sarcoma was previously identified as an independent prognostic factor (7), but more recent studies have not found an association potentially due to improved therapeutic regimens (8). Alternatively, in alveolar rhabdomyosarcoma, translocation-negative tumors have better outcomes than translocation-positive tumors despite histologic appearance (9). Although genomic profiling has increased our understanding of sarcomageneis, to date no routine therapies have been identified to target specific translocations in sarcoma. However, the success of the targeted therapy imatinib in the treatment of gastrointestinal stromal tumors, directed at an oncogenic mutation in KIT, suggests a role for subtype-specific molecular therapies.
SOFT TISSUE SARCOMA
Soft tissues include fibrous, adipose, and vascular structures as well as muscles and tendons and are mesenchymal in origin. Soft tissue sarcomas are a heterogeneous group encompassing approximately 60 different subtypes, based on their resemblance to normal tissues rather than the tissue of origin.
Soft tissue sarcomas can occur in any anatomic region. The majority of soft tissue sarcomas arise from the extremities (60%), followed by the trunk (30%) and the head and neck region (10%). The most common presenting symptom is a soft tissue mass or swelling. Pain is reported by only about one-third of patients at presentation. Therefore, because of the lack of symptoms, there is often a delay in the diagnosis. Patients with a soft tissue mass that is increasing in size, a mass >5 cm, or a mass that is deep to deep fascia, regardless of pain, should be referred for evaluation of a suspected soft tissue sarcoma (10).
Evaluation of a suspected soft tissue sarcoma begins with a comprehensive history and physical examination. Imaging evaluation is dependent on the site of disease. For soft tissue tumors of the extremities, head and neck, and pelvis, magnetic resonance imaging (MRI) is preferred. Soft tissue sarcomas found in the retroperitoneum and abdomen are generally best evaluated by computed tomography (CT). Positron emission tomography (PET) imaging can distinguish histologic grade to a degree based on tumor standardized uptake values (SUV) (11). For both soft tissue sarcomas and bone sarcomas, PET imaging has a predictive role in determining response to chemotherapy and targeted agents such as imatinib, but it has also been shown to predict survival after initial cycle of therapy among patients receiving neoadjuvant chemotherapy for high-grade soft tissue sarcoma (12,13).
A biopsy is essential to diagnosis, and the method of biopsy chosen should be the least invasive technique available to make a definitive diagnosis. A core-needle biopsy is sufficient; however, multiple cores should be obtained to improve diagnostic yield. If an open biopsy is performed, it should be planned such that the biopsy tract can be removed at the time of definitive surgical resection to reduce the risk of seeding and recurrence (14). An excisional biopsy may be used for small or superficial lesions; however, careful examination of margins and planning of the orientation of the resection should always be performed. Fine-needle aspiration (FNA) is not recommended but can be useful in confirming recurrence, assuming that an experienced sarcoma cytopathologist is available (15).
Sarcomas are classified primarily according to their tissue appearance, histologic grade, and sometimes the cell of origin. This can be difficult, as approximately 60 different histologic types of soft tissue sarcoma are recognized (Table 42-2). Histologic diagnosis is classified by the updated 2013 World Health Organization criteria (16).
| Sarcomas of adipose tissue |
| Liposarcoma |
| Atypical lipomatous tumor |
| Myxoid liposarcoma |
| Cellular myxoid liposarcoma |
| Round cell liposarcoma |
| Dedifferentiated liposarcoma |
| Pleomorphic liposarcoma |
| Sarcomas of peripheral nervous tissue |
| Malignant peripheral nerve sheath tumor |
| (Malignant schwannoma, neurofibrosarcoma, neurogenic sarcoma) |
| Sarcomas of smooth muscle |
| Leiomyosarcoma |
| Sarcomas of fibrous tissue |
| Desmoid fibromatosis |
| Dermatofibrosarcoma protuberans |
| Low-grade fibromyxoid sarcoma |
| Fibrosarcoma |
| Malignant fibrous histiocytoma (MFH) |
| Sarcomas of blood vessels and lymphatics |
| Epithelioid hemangioendothelioma |
| Hemangiopericytoma |
| Angiosarcoma/lymphangiosarcoma |
| Sarcomas of skeletal muscle |
| Embryonal rhabdomyosarcoma |
| Alveolar rhabdomyosarcoma |
| Pleomorphic rhabdomyosarcoma |
| Sarcomas of unknown origin |
| Synovial sarcoma |
| Monophasic |
| Biphasic |
| Alveolar soft tissue sarcoma |
| Epithelioid sarcoma |
| Unclassified sarcoma |
| Extraskeletal osteosarcoma |
| Extraskeletal chondrosarcoma |
| Extraskeletal Ewing sarcoma (PNET) |
| Soft tissue tumors of melanocytic tissue |
| Melanoma of soft tissue or clear cell sarcoma |
Pathologists often separate sarcomas into three histologic grades using the Federation Nationale des Centres de Lutte Contre le Cancer (FNCLCC) grading system. Biological aggressiveness can be predicted based on histologic grade, and this spectrum varies among the histologic subtypes of sarcoma (17). Immunohistochemistry, cytogenetics, and molecular pathology can aid in making a diagnosis; however, some poorly differentiated spindle cell neoplasms cannot be categorized further.
The American Joint Committee on Cancer (AJCC) staging system may be used for soft tissue sarcomas (Table 42-3) (18). All soft tissue sarcoma subtypes are included except Kaposi sarcoma, dermatofibrosarcoma protuberans, infantile fibrosarcoma, and angiosarcoma. This system is designed to classify tumors of the extremities, trunk, head and neck, and retroperitoneum, but it was not designed for evaluation of sarcomas of the gastrointestinal (GI) tract. This system has its limitations because anatomic site and certain histologic subtypes (eg, small cell histologies) that are known to influence outcome are not taken into account (19).
| Primary tumor (T) | ||||||
| TX | Primary tumor cannot be assessed | |||||
| T0 | No evidence of primary tumor | |||||
| T1 | Tumor 5 cm or less in greatest dimension | |||||
| T1a superficial tumora | ||||||
| T1b deep tumora | ||||||
| T2 | Tumor more than 5 cm in greatest dimension | |||||
| T2a superficial tumor | ||||||
| T2b deep tumor | ||||||
| Regional lymph nodes (N) | ||||||
| NX | Regional lymph nodes cannot be assessed | |||||
| N0 | No regional lymph node metastasis | |||||
| N1 | Regional lymph node metastasis | |||||
| Distant metastasis (M) | ||||||
| MX | Distant metastasis cannot be assessed | |||||
| M0 | No distant metastasis | |||||
| M1 | Distant metastasis | |||||
| Histologic grade (G) | ||||||
| GX | Grade cannot be assessed | |||||
| G1 | Well differentiated | |||||
| G2 | Moderately differentiated | |||||
| G3 | Poorly differentiated | |||||
| G4 | Poorly differentiated or undifferentiated (four-tiered systems only) | |||||
| Stage grouping | ||||||
| Stage I | T1a, 1b, 2a, 2b | N0 | M0 | G1-2 | G1 | Low |
| Stage II | T1a, 1b, 2a | N0 | M0 | G3-4 | G2-3 | High |
| Stage III | T2b | N0 | M0 | G3-4 | G2-3 | High |
| Stage IV | Any T | N1 | M0 | Any G | Any G | High or low |
Several clinicopathologic factors are important for treatment planning and prognosis assessment. These form the basis for the AJCC classification system and include tumor grade, size of the primary tumor, depth of invasion, and extent of disease (17). High-risk features for local recurrence or distant metastases are high-grade lesions, primary tumor >5 cm, and deep tumor location. Approximately 50% of patients with intermediate- and high-grade soft tissue sarcoma will develop metastatic disease requiring systemic therapy (20). The 5-year overall survival for soft tissue sarcoma is around 50%, with local control and distant disease being the key determinants (21).
Treatment of sarcoma requires a multidisciplinary approach with experienced medical, surgical, and radiation oncologists, pathologists, and radiologists. An improved understanding of soft tissue sarcoma subtypes in regard to natural history, response to chemotherapy, and potential for targeted therapies has led to more subtype-specific treatment according to individual histology.
For local disease, surgical resection is the mainstay of treatment. Sarcomas tend to expand and compress tissue planes, which produce a pseudo-capsule comprising normal tissue interlaced with tumor tissue. Wide local excision with a margin of normal tissue surrounding the tumor is associated with lower local recurrence rates of approximately 10% to 30% (17). The ideal surgical margins should be 2 to 3 cm without tumor involvement. If positive margins are confirmed by pathology, re-excision to obtain negative margins is important when feasible to improve local control and relapse-free survival. For patients with borderline resectable tumors, consideration should be given to neoadjuvant therapy depending on the tumor histology and patient’s performance status.
Adult sarcomas have a less than 4% prevalence of lymph node metastases (22). For this reason, routine regional lymph node dissection is often not required. However, patients with synovial sarcoma, clear cell sarcoma, rhabdomyosarcoma, angiosarcoma, and epithelioid sarcomas have a higher incidence of lymph node metastases and should be evaluated closely for lymphadenopathy.
Improved surgical techniques and multimodality treatment have resulted in a decrease in radical resection of extremity tumors with a corresponding rise in limb-sparing procedures combining wide local resection with preoperative or postoperative chemotherapy and radiotherapy. Approximately 90% of patients with localized sarcomas of the extremities can safely undergo limb-sparing procedures to preserve limb function and adequately maintain local control (23). A study conducted at the National Cancer Institute (NCI) showed no survival advantage to amputation over limb-sparing surgery with postoperative radiation (24).
Although radiation is not effective for the treatment of gross disease, it has been a useful adjunct to surgery in the treatment of microscopic local disease and for palliation of symptoms. Radiation therapy is commonly used in the preoperative or postoperative adjuvant setting. Because there are pros and cons as to the timing of radiation therapy, this topic remains controversial; appropriate discussion between radiation oncologists, medical oncologists, and surgeons is required in planning the treatment of each patient.
Preoperative radiation has several advantages over postoperative radiation, including smaller radiation portals, conversion to a limb-sparing procedure, reduction of the extent of the surgical procedure, and lower radiation doses, which can be used because there are theoretically fewer radio-resistant hypoxic cells within the tumor and surgical removal can supplement the boost (25). However, preoperative radiotherapy may lead to difficulty in assessing pathologic responses to preoperative chemotherapy and may also contribute to delayed wound healing. Several studies have shown improved local control rates with preoperative radiation, especially with larger tumors that were initially considered unresectable (26). The modality of choice is external beam radiotherapy (EBRT), and a dose of 50 Gy or more is often required to obtain local control. At these dose levels, the entire circumference of the extremity must not be irradiated in order to avoid lymphedema. A period of 4 to 6 weeks is needed following preoperative radiation to prevent wound complications. Following the surgical resection, close or positive margins could be treated with a radiation boost if feasible. Brachytherapy, EBRT, or intraoperative radiotherapy can be used by experienced clinicians in appropriate situations (27).
Postoperative radiation therapy should be considered in patients with high-grade soft tissue sarcomas of the extremities with positive microscopic margins (<1 mm from the inked margin). In this setting, adjuvant radiation improved the 5-year local control rate compared to the no RT group (74% vs 56%; P = .01) (28). More recently, adjuvant intensity-modulated radiation therapy has been shown to reduce local recurrence as compared to conventional EBRT for primary soft tissue sarcoma of the extremity (hazard ratio [HR], 0.46; P = .02) (29). The interval of time between surgery and initiation of radiation therapy is a controversial but legitimate concern. The most recent soft tissue sarcoma guidelines issued by the National Comprehensive Cancer Network (NCCN) suggest the interval should be no greater than 6 weeks (30).
Radiation therapy is occasionally used as the sole treatment modality for palliation for some patients with soft tissue sarcomas. These patients are often those who have unresectable disease or who are not appropriate candidates for surgery and/or chemotherapy. There have been reports of 5-year survival rates ranging from 25% to 40% with radiation therapy alone and of local control rates of approximately 30%, depending on the primary tumor’s size and biology (31).
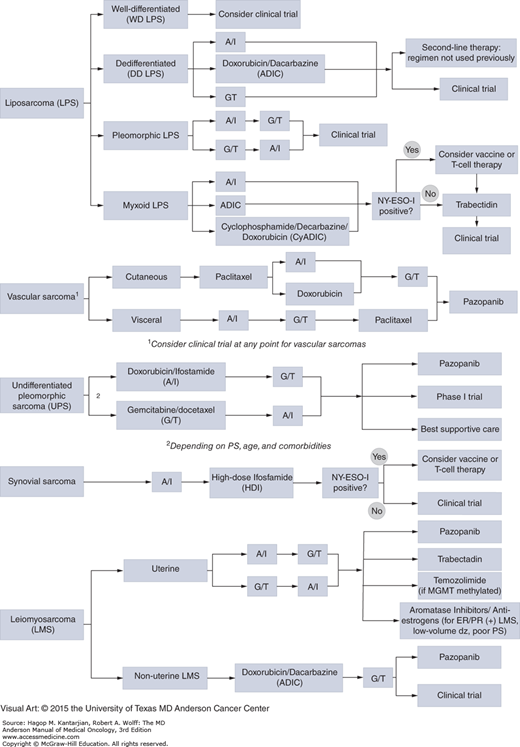
Systemic therapy for soft tissue sarcomas is primarily used in the metastatic/advanced disease setting, whereas the role of chemotherapy in the neoadjuvant and adjuvant setting is less well established. Treatment relies primarily upon conventional chemotherapy agents, which are largely unchanged over the past two decades. In general, tumors with a higher grade are more likely to responds to chemotherapy; however, chemosensitivity varies based on histologic subtype (Table 42-4) (32). An understanding of chemosensitivity and molecular aberrations based on subtype has led to histology-driven treatment algorithms for specific soft tissue sarcomas such as leiomyosarcoma, myxoid liposarcoma, and angiosarcomas (Fig. 42-1). This approach is particularly important in considering targeted therapies for specific subtypes that are considered chemoresistant such as alveolar soft parts sarcoma.
FIGURE 42-1
Approach to systemic therapies for advanced soft tissue sarcomas. A/I doxorubicin/ifosfamide; ADIC doxorubicin, dacarbazine; CyADIC, cyclophosphamide, doxorubicin, dacarbazine; ER, estrogen receptor; G/T gemcitabine/docetaxel; MGMT, O6-methylguanine-DNA methyltransferase; PR, progesterone receptor; PS, performance status.
| Relative Chemosensitivity | Example |
|---|---|
| Highly sensitive (chemotherapy standard of care in management) | Ewing sarcoma family of tumors/PNET Embryonal and alveolar rhabdomyosarcoma |
| Sensitive to chemotherapy | Synovial sarcoma Small cell sarcoma Myxoid/round cell liposarcoma Uterine leiomyosarcoma |
| Moderate sensitivity to chemotherapy | Pleomorphic liposarcoma Myxofibrosarcoma Epithelioid sarcoma Pleomorphic rhabdomyosarcoma Leiomyosarcoma Malignant peripheral nerve sheath tumor (MPNST) Angiosarcoma Desmoplastic small round cell tumor (DSRCT) |
| Insensitive to chemotherapy | Dedifferentiated liposarcoma Clear cell sarcoma Endometrial stromal sarcoma |
| Chemoresistant (chemotherapy risk clearly outweighs benefit) | Gastrointestinal stromal tumor (GIST) Alveolar soft parts sarcoma (ASPS) Extraskeletal myxoid chondrosarcoma |
The two most active agents in the treatment of soft tissue sarcoma are doxorubicin and ifosfamide. Doxorubicin is most active at doses of ≥75 mg/m2, with single-agent response rates of approximately 20% to 35% (33). Ifosfamide has been shown to produce single-agent response rates similar to those of single-agent doxorubicin when used at doses of 10 g/m2 or higher (34). Ifosfamide has also been shown to have greater efficacy when administered as a 2- to 3-hour infusion as opposed to a 24-hour infusion (34,35). Studies have shown that both doxorubicin and ifosfamide also exhibit a positive dose-response curve (33,34). The response rate in soft tissue sarcoma patients whose disease failed doxorubicin-based therapy and who then received high-dose ifosfamide as a single agent was 29% (34). Therefore, high-dose ifosfamide as a single agent at doses of 14 g/m2 is sometimes used as a salvage regimen at the University of Texas MD Anderson Cancer Center (MDACC) for selected histologies.
Combination therapy with dose-intense doxorubicin and ifosfamide has been shown to improve response rates and progression-free survival (PFS) and possibly overall survival (36). The combination of doxorubicin (75 or 90 mg/m2) and ifosfamide (at 10 g/m2) was evaluated at MDACC in patients with soft tissue sarcomas and demonstrated a 75% response rate (95% confidence interval [CI], 59%-71%; complete response [CR], 12%) in patients with primary tumors of the extremities and a 68% response rate (95% CI, 56%-80%; CR, 12%) in patients with primary disease at any site (37). The response rates according to histology were as follows: malignant fibrous histiocytoma, 69%; synovial sarcoma, 88%; unclassified sarcomas, 60%; non-GI leiomyosarcomas, 50%; liposarcomas, 56%; angiosarcomas, 83%; and neurogenic sarcomas, 40%; other miscellaneous histologies demonstrated objective response rates of 45% (37). In the large randomized phase III European Organization for Research and Treatment of Cancer (EORTC) 62012 trial comparing single-agent doxorubicin with doxorubicin in combination with ifosfamide, the combination group had a significantly higher response rate (26% vs 14%) and increased median PFS (7.4 vs 4.6 months) but also had an increase it grade 3 and 4 toxicities. Although overall survival at 1 year was increased in the combination group (60% vs 51%, P = .076), this failed to meet statistical significance (38). At our center, we continue to use combination dose-intense doxorubicin and ifosfamide in appropriately selected patients and preferentially in the neoadjuvant setting for large (≥5 cm), high-grade, resectable soft tissue sarcomas.
Dacarbazine has activity as a single agent, with response rates of 10% to 15%. The three-drug regimen MAID (mesna, doxorubicin [Adriamycin], ifosfamide, dacarbazine) has been studied and has shown response rates varying from 25% to 47% (39). When the MAID regimen was studied at MDACC, significant toxicities related to the addition of dacarbazine were seen (40). The combination of doxorubicin and dacarbazine (ADIC) is often used in extrauterine leiomyosarcoma or as a second-line regimen in other soft tissue sarcomas. In patients with advanced/metastatic leiomyosarcoma or liposarcoma treated with ADIC as first-line therapy, Response Evaluation Criteria in Solid Tumors (RECIST) response rates of 57% and 40% were observed, respectively (41).
Gemcitabine alone or in combination with docetaxel is frequently used for the treatment of advanced, recurrent, or metastatic disease once patients fail doxorubicin- and ifosfamide-based therapy or in patients who may not tolerate intensive chemotherapy. An initial phase II study using gemcitabine as a single agent demonstrated a response rate of 18% (95% CI, 7%-29%), including many pretreated patients (42). The synergistic effect of docetaxel when added to gemcitabine was evaluated in a randomized phase II study, SARC002 (43). By RECIST, response rates for the gemcitabine-docetaxel arm and gemcitabine arm was 16% and 8%, respectively. Furthermore, an improvement in median PFS (6.2 vs 3.0 months) and overall survival (17.9 vs 11.5 months) was noted in the gemcitabine-docetaxel arm compared to the gemcitabine arm. The two histologies most responsive to the gemcitabine-docetaxel arm were leiomyosarcoma and high-grade undifferentiated pleomorphic sarcoma.
Trabectedin, a novel antitumor compound initially isolated from extracts of sea squirt Ecteinascidia turbinata through the NCI drug screening program in the 1960s, has shown activity in the second-line treatment of soft tissue sarcomas. The mechanism of action of trabectedin is complex but is thought to involve displacement of transcription factors from their promoter (44). Additionally, sensitivity of myxoid liposarcoma, a translocation-related soft tissue sarcoma, has been shown to correlate with expression of the FUS-DDIT3 fusion gene (45). Taken together, these factors suggest a role for trabectedin in translocation-related sarcomas and pose a potential mechanism of action. In a single-arm phase II trial of trabectedin as second- or third-line therapy in advanced soft tissue sarcoma, the overall response rate was 8% (46). However, many patients demonstrated prolonged disease stabilization, with 26% with stable disease >6 months with minimal toxicities. This benefit was greatest in leiomyosarcomas and translocation-related sarcomas. A retrospective review of eight phase II trials of trabectedin in translocation-related soft tissue sarcomas demonstrated encouraging results in regard to disease control, with greatest activity in myxoid liposarcoma (47). This has led to a current phase III trial of first-line therapy with trabectedin versus doxorubicin-based chemotherapy in translocation-related sarcomas. Currently, trabectedin is approved in Europe for second-line treatment of soft tissue sarcoma and has been granted orphan drug status by the US Food and Drug Administration.
The goals of chemotherapy in the treatment of high-risk local disease are to eradicate micrometastasis, decrease risk of local recurrence, and downsize tumors to facilitate either limb-sparing procedures for extremity tumors or resection for tumors initially deemed unresectable (Fig. 42-2). At MDACC, preoperative chemotherapy is preferred in patients with high-risk (>5 cm or high-grade) tumors and in patients who are considered borderline resectable with chemosensitive soft tissue sarcoma subtypes.

Postoperative chemotherapy and its benefits continue to be controverted as trials of adjuvant therapy have yielded conflicting results. In the most recent update to the Sarcoma Meta-Analysis Collaboration (SMAC) conducted in 2008, the benefit of adjuvant chemotherapy was analyzed among 1,953 patients with soft tissue sarcoma across 18 trials (48). This update incorporated five trials evaluating doxorubicin and ifosfamide in combination, a regimen not previously represented in the initial SMAC analysis. This updated meta-analysis detected favorable odds ratios (ORs) of local recurrence and distant recurrence for chemotherapy. Although the absolute risk reduction (ARR) in distant recurrence with adjuvant doxorubicin-based chemotherapy for all studies was 9% (95% CI, 5%-14%; P = .000), the ARR with adjuvant doxorubicin-ifosfamide chemotherapy was 10% (95% CI, 1%-19%; P = .03) (48). By pooling the data, the number needed to treat (NNT) to prevent distant recurrence was 12. Although a survival benefit was not noted with single-agent doxorubicin, a statistically significant survival advantage was observed with the doxorubicin-ifosfamide combination. The OR for overall survival in the doxorubicin-ifosfamide cohort was 0.56 (95% CI, 0.36-0.85; P = .01). Combining all trials in the meta-analysis, the NNT to prevent one death was 17. A recent randomized controlled trial of adjuvant therapy with doxorubicin 75 mg/m2 and ifosfamide 5 g/m2 in patients with intermediate- or high-grade STS failed to demonstrate a benefit in overall survival (HR, 0.94; P = .72) or relapse-free survival (HR, 0.91; P = .51) (49). Although the data regarding adjuvant therapy are conflicting, within our institution, we continue to offer adjuvant therapy with doxorubicin in combination with ifosfamide to healthy patients with intact organ function who have high-risk disease (tumor size >5 cm, high-grade histology, and deep soft tissue involvement).
As in other tumor types, increased knowledge of cancer genomics and identification of oncogenic driver mutations in soft tissue sarcomas have led to much enthusiasm and investigation of molecular-based targeted therapies. A comprehensive review of targeted therapies under development for soft tissue sarcoma is beyond the scope of this chapter, and therefore, the focus will be on currently approved therapies. Targeting cKIT with the tyrosine kinase inhibitor (TKI) imatinib in gastrointestinal stromal tumors (GISTs) is perhaps the best-known and most successful example in sarcoma. Although targeted agents have shown promise in specific histologies, the multitargeted TKI pazopanib has shown activity across multiple subtypes of soft tissue sarcomas. Pazopanib is a small-molecule inhibitor with activity against VEGF1-3, PDGFRA, PDGFRB, and KIT. A phase II trial of pazopanib in advanced soft tissue sarcoma evaluating 12-week PFS as the primary end point showed benefit in leiomyosarcoma (44%), synovial sarcoma (49%), and other nonlipomatous soft tissue sarcoma (39%) (50). Subsequently, a placebo-controlled phase III trial of pazopanib in metastatic soft tissue sarcoma demonstrated a low response rate (partial response [PR], 6%) but significant improvement in PFS (4.6 months vs 1.6 months with placebo; HR, 0.31; P < .0001) (51). In a multivariate Cox model, favorable prognostic factors in patients treated with pazopanib were good performance status and low or intermediate tumor grade. Additional targeted therapies in soft tissue sarcoma are primarily being developed and studied in specific soft tissue sarcoma subtypes.
Patients with metastatic disease involving multiple organs are generally incurable and considered appropriate for palliative systemic therapy as described earlier. The subset of patients with lung-only metastatic disease, especially with a greater than 12-month disease-free interval, have a favorable biology and prognosis and therefore should be considered for resection if feasible. This approach results in 3- to 5-year survival of up to 20%. Chemotherapy is the mainstay of therapy for patients with metastatic disease, although surgical resection of residual disease to render patients free of gross disease is often pursued. The sequencing of chemotherapy is similar to that of isolated local disease. In a study conducted at MDACC, patients with metastatic disease showed a 57% response rate to doxorubicin (75-90 mg/m2) and ifosfamide (10 g/m2) (37). If patients fail this regimen, the choice of treatment depends on the histology of the tumor and the performance status of the patient.
Vascular sarcomas are tumors that originate from or differentiate toward the endothelium with varying malignant potential. Although epithelioid hemangioendotheliomas have an intermediate malignant potential and indolent clinical course, angiosarcomas, at the other end of the spectrum, have a highly malignant biologic behavior with early propensity for distant metastasis and dismal outcomes. These tumors also differ in their response to chemotherapy and targeted therapy and, therefore, are discussed separately below.
Epithelioid hemangioendotheliomas (EHE) are considered to be of intermediate malignant potential with development of metastasis and recurrence. They typically are associated with a blood vessel, usually a medium sized or large vein. Epithelioid hemangioendothelioma most commonly occurs in the soft tissues, but liver, lung, and bone may be sites of primary involvement. In over 42% of patients with hepatic EHE, symptoms are often absent and the lesions are discovered incidentally. Some patients experience constitutional symptoms such as fatigue, anorexia, nauseam and poor exercise tolerance. Most cases of EHE affecting soft tissues are localized, whereas multifocality is more common with EHE involving liver or lung, and these patients develop metastatic disease during the course of their illness. Multifocal or metastatic disease does not equate to mortality, and many patients can survive long term with metastatic disease. Sixty-three percent of patients with liver EHE and less than half of patients with metastatic EHE of soft tissues die from their disease.
Localized EHE of soft tissue should be treated with surgical resection with adequate margins. Following resection, these tumors can recur locally in about 12% of patients (52). Preoperative radiation therapy should be considered in patients where good margins are unlikely, and postoperative radiation therapy should be considered in cases where the margins are positive and no preoperative radiation was administered. Localized EHE does not require the use of chemotherapy or targeted therapies.
Metastatic EHE of soft tissue may be followed without therapy until there is evidence of progressive disease on serial imaging over a 3-month period. When systemic therapy is needed, conventional chemotherapy and antiangiogenic therapy may be considered. Systemic therapy options include gemcitabine, taxanes, and doxorubicin. Targeted therapy with bevacizumab (PR, 29%; stable disease, 57%; and progressive disease, 14%) (53), sorafenib (30.7% without progression at 9 months) (54), and interferon α-2b (55) has been reported to have utility in patients with metastatic EHE.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


