INTRODUCTION
Myelodysplastic syndromes (MDS) refer to a group of hematopoietic disorders characterized by ineffective hematopoiesis and increased risk of transformation to acute myelogenous leukemia (AML). Most patients with MDS succumb to causes related to the disease. The median age of patients with MDS is 70 to 75 years. It is likely that environmental factors play an important role in the pathogenesis of this disease. Myelodysplastic syndromes are classified according to the World Health Organization (WHO) criteria, and a number of prognostic scores can be used to calculate survival and risk of transformation. Cytogenetic, genomic, and epigenetic alterations are common in MDS and help in the prediction of prognosis and potentially in the selection of therapy. Over the last decade, we have witnessed significant improvements in supportive care and therapeutic modalities for patients with MDS. These include growth factors, immune modulatory agents (lenalidomide), and hypomethylating agents (5-azacitidine and decitabine). We also better understand patient subgroups, such as those with hypomethylating failure disease. In this chapter, we summarize our knowledge of MDS and the treatment approach we use at MD Anderson Cancer Center.
THE MD ANDERSON APPROACH TO THE PATIENT WITH MDS
Every year, approximately 350 to 400 patients are referred to our center with a diagnosis of MDS. Nearly 20% of patients referred with a diagnosis of MDS receive a different diagnosis in our center. In most instances, the final diagnosis is that of AML or a form of higher-risk MDS. Other benign and malignant conditions can also be observed. In a study of 915 patients referred between 2005 and 2009 and using very strict criteria, 12% were reclassified when initially evaluated here (1). This justifies our practice to repeat a confirmatory bone marrow aspiration and biopsy at the time of initial MDS evaluation at MD Anderson.
Once the diagnosis is confirmed, the next important step is to calculate the “risk” of the patient. Most clinicians and investigators still use the International Prognostic Scoring System (IPSS) (2) score to perform such analysis, but newer potentially more precise models, such as the Revised International Prognostic Scoring System (IPSS-R), have been developed (3,4,5). In general, patients with low or intermediate-1 risk by the IPSS or those with less than 10% blasts in the bone marrow are considered as having “lower” risk disease, whereas those with excess blasts or intermediate-2 or high-risk disease are considered as having “higher” risk disease.
Patients with lower risk disease can be candidates for a wide range of interventions, depending on their specific characteristics and transfusion needs. Patients with minimal cytopenias, who are transfusion independent, who have a low percentage of blasts in the bone marrow, and have diploid cytogenetics are more frequently observed, as their 4-year survival is close to 80% (4). At the end of the spectrum, older patients with significant cytopenias and transfusion needs can have very poor prognosis, particularly if their cytogenetics are abnormal (4). The median survival of these patients is less than 12 months; around 60% to 70% of patients with MDS are in this category, but there are few interventions known to alter the natural history of these patients. Transfusion and growth factor support are usually started. Interventions such as lenalidomide have significant activity in improving red cell counts in patients with deletion of chromosome 5 (6) but are significantly less active in patients without this alteration (7). The role of the hypomethylating agents 5-azacitidine or decitabine is less clear in this situation, although they are frequently used. Allogeneic stem cell transplantation (alloSCT) is not frequently used up front in patients with lower risk disease (8). It is currently accepted that delaying transplantation until the time of progression is associated with longer survival even if transplant outcomes are poorer when performed at that time. A new subset of patients has emerged that is constituted by patients with lower risk disease but with hypomethylating failure. We recently described their natural history (9). New investigational strategies are needed for this subset of patients.
Treatment decisions are relatively simpler for patients with higher risk MDS. The data with the hypomethylating agents indicate that treatment with these agents improves survival significantly when compared to supportive care or low-dose chemotherapy approaches. The optimal approach for younger (those less than 60 to 65 years) patients with MDS is unclear. These patients can be treated with hypomethylating agents or an AML-like induction therapy or can be considered for up-front alloSCT. No study has compared these treatments in younger patients. An approach followed by our group is to stratify patients based on cytogenetics. Younger patients with normal karyotype are usually offered induction therapy with an AML-like approach followed when possible by alloSCT. In contrast, younger patients with abnormal karyotypes are offered hypomethylating agent-based therapy followed by alloSCT. It is not our routine to proceed with transplant up front in patients with excess blasts. Older patients benefit significantly from the use of hypomethylating agents, and there is basically no upper age limit that may contraindicate their use (10). Finally, the group of patients with higher risk disease and hypomethylating failure constitute a major medical need (11).
A comprehensive review of current knowledge in MDS is provided next. Current areas of intense research are the development of newer forms of therapy for patients with newly diagnosed disease and strategies for patients who have relapsed or not responded to hypomethylating agent-based therapy.
EPIDEMIOLOGY AND ETIOLOGY
The incidence of MDS increases with age. Most patients diagnosed with this condition are more than 60 years old; the median age at diagnosis is 75 years (12). The incidence is higher in males than females, with a 2:1 ratio (12). The incidence in the United States is 30 to 35 individuals per million per year with a relative yearly increase in the reported incidence, probably related to increase awareness of the disease and reporting efforts (13).
The risk of developing MDS is related to the individual’s racial background. In the United States, the incidence is highest in the white population (12). Patients with MDS from Asia present at a younger age (14). The underlying cause of this phenomenon is not known but may reflect genetic differences between different racial groups. Asian patients have a similar frequency of karyotype abnormality as European and American cohorts, although they may have less frequent alterations of chromosomes 5 and 7 (14,15,16). The combination of younger age at diagnosis and lack of chromosome 7 alterations can explain the longer survival observed in patients from Asia.
There is no known cause of MDS. Genetic or environmental risk factors may contribute to MDS. Genetic syndromes, such as Down syndrome, Bloom syndrome, and Fanconi anemia, are associated with an increased risk of MDS, which often presents earlier in life (17,18). Genetic polymorphisms that influence the activity of enzymes responsible for metabolizing toxic chemicals or chemotherapy drugs may influence an individual’s predisposition to MDS. Polymorphisms have been described in the cytochrome p450 3A, glutathione-S-transferase, and NAD(P)H quinine oxidoreductase enzyme systems that increase the risk of developing myeloid malignancy (19,20,21).
Environmental agents may contribute to the development of MDS by causing toxic damage to hematopoietic stem cells. A causal relationship between occupational exposures to benzene and radiation and the development of myeloid malignancy has been demonstrated (22). Exposure to organic solvents and pesticides has also been implicated in the development of MDS (23,24,25). There is no correlation between MDS and socioeconomic status (24).
The most significant risk factor for the development of MDS is previous exposure to chemotherapy or radiotherapy used to treat other cancers. Treatment-related MDS (t-MDS) constitutes a minority of MDS diagnoses but may be increasing in prevalence with improved survival rates after successful cancer therapies for tumors. Usually, t-MDS presents 5 to 6 years after initial cancer treatment and generally has a poor prognosis (26). Patients treated for lymphoma are at risk of this long-term complication (27). Patients who undergo autologous hematopoietic stem cell transplantation have a higher risk (10%-15%) of developing treatment-related MDS or AML, with incidence rates in some centers of up to 10% (28). In our experience with t-MDS at MD Anderson in 281 patients, most of the risk was associated with complex cytogenetics or presence and alteration of chromosome 7 (29).
CLINICAL AND LABORATORY FEATURES
Most patients with MDS are diagnosed incidentally during routine complete blood cell count (CBC) analysis or because of nonspecific symptoms. Anemia is the most common cytopenia in MDS and is associated with fatigue. A lower percentage of patients presents with bleeding or bruising secondary to thrombocytopenia or with infections related to neutropenia. Physical examination is often normal. Hepatosplenomegaly may be present in patients with chronic myelomonocytic leukemia (CMML) or overlap myeloproliferative neoplasms. A change in the severity of cytopenia or rapid worsening of symptoms may indicate disease transformation. Patients suspected to have MDS transformation require prompt investigation because 20% to 30% of patients will develop acute leukemia throughout their disease course (2).
Initial assessment should include a CBC, reticulocyte count, and serum chemistry, including evaluation of B12 and folate, iron studies (ferritin), and erythropoietin level examination. Other causes of cytopenias or MDS-mimicking syndromes (eg, HIV [human immunodeficiency virus], other infections, autoimmune disorders, or copper deficiency) should be ruled out through appropriate tests. A bone marrow aspirate and biopsy with samples taken for an iron stain and cytogenetic studies are required. Morphological assessment of the disease is still required for MDS. Cytogenetic studies may confirm the presence of clonal hematopoiesis and provide additional important prognostic information. Analysis of specific gene mutations, such as TET2, DNMT3A, ASXL1, TP53, splicing factors, NRAS, FLT-3, IDH1, IDH2, and JAK2 may improve our prognostic and predictive evaluation and may in time allow for the use of targeted interventions using selective inhibitors (eg, FLT3, JAK2, or IDH1/2 inhibitors) or allow earlier consideration of alloSCT. However, it should also be noted that these molecular abnormalities (30) may be present in older individuals with or without cytopenias and may not necessarily point to an MDS diagnosis (31,32). In general, no patient should be diagnosed as having MDS without knowledge of the clinical and drug history or while on growth factor therapy, including erythropoietin.
MORPHOLOGICAL FEATURES
Morphological classification of MDS is based on a 500-cell differential count on the bone marrow aspirate and leukocyte differential performed on the blood smear (33). Table 5-1 shows the different subsets of MDS under the newly revised WHO MDS classification. This analysis determines the percentage of blasts present in the blood and bone marrow and provides an assessment of the number of myeloid lineages involved in the dysplastic process, and the iron stain determines the presence and number of ring sideroblasts (Fig. 5-1) (33).
|
FIGURE 5-1
Morphological features of peripheral blood and bone marrow in the myelodysplastic syndromes. A. Peripheral blood film from a patient with refractory anemia with excess blasts-1. The erythrocytes show hypochromasia, anisocytosis, and macroovalocytes. There is also an occasional blast (center). B. Peripheral blood film from a patient with refractory cytopenia with multilineage dysplasia demonstrating pseudo–Pelger-Huet cell (center) with hypercondensed chromatin and bilobed nuclei and hypogranular cytoplasm. C. Dysplastic small megakaryocytes, some with monolobated or with separated nuclei and mature granular cytoplasm in the bone marrow aspirate from a patient with refractory anemia with excess blasts. D. Increased blasts, dysgranulopoiesis, and dyserythropoiesis in the bone marrow aspirate from a patient with refractory anemia with excess blasts. E. Ring sideroblasts and Pappenheimer bodies from a patient with refractory anemia with ring sideroblasts. F. Hypercellular (100%) bone marrow biopsy with increased immature cells and dysplastic megakaryocytes in a 70-year-old male with refractory anemia with excess blasts.
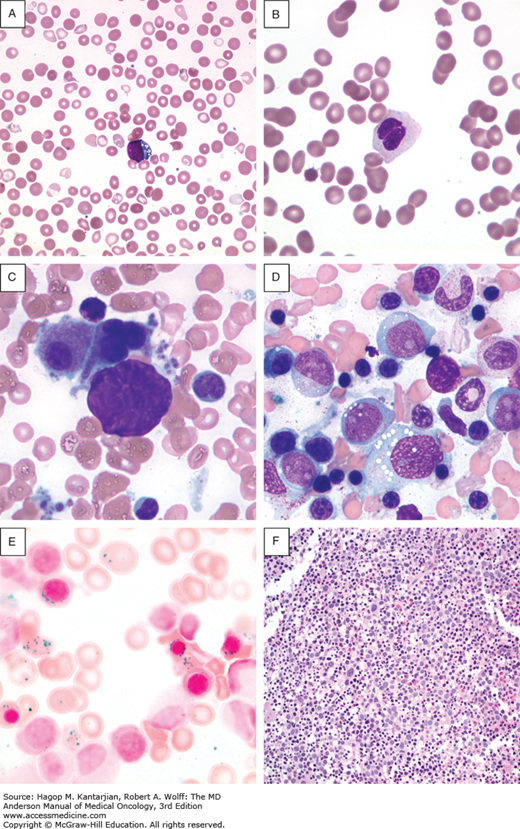
Blood cell abnormalities on the peripheral blood smear are variable (33) (see Fig. 5-1). Red cells may be macrocytic and frequently display anisopoikilocytosis. Polychromasia or basophilic stippling may be present. Dysplastic granulocytes may show abnormal folding of the nucleus, and cytoplasmic granules are often reduced or absent. Platelets are of variable size and may also be hypogranular. The presence of circulating blast cells or an excess of monocytes is important for the classification of high-risk MDS and CMML, respectively.
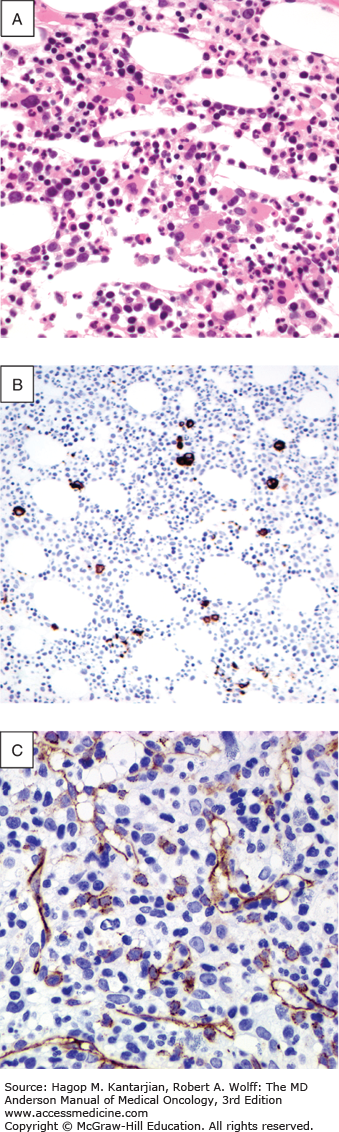
Definitive diagnosis requires a bone marrow aspirate and biopsy. The bone marrow is usually normocellular or hypercellular, reflecting that hematopoiesis is ineffective. Abnormal maturation of hematopoietic cells results in a variable proportion of myeloblasts that are significantly increased in the more aggressive MDS. Morphological abnormalities found in the nucleus of erythroblasts include nuclear budding, internuclear bridging, karyorrhexis, multinuclearity, and megaloblastoid changes (see Fig. 5-1). Cytoplasmic features include the presence of ring sideroblasts and abnormal vacuolization. Abnormal or absent granulation is a common feature of dysplastic granulocyte series. Aberrant nuclear folding of the neutrophil precursor can produce a dysplastic bilobed nucleus, the pseudo–Pelger-Huet anomaly. Megakaryocytes may have a very variable morphology, and a small dysplastic form called the micromegakaryocyte is a typical finding. A normal megakaryocyte has a polyploid nucleus that can be altered with dysplasia to produce hypolobulation or nuclei that are dispersed throughout the cell. The bone marrow biopsy provides the best assessment of the overall cellularity and allows examination of the architecture of the marrow and surrounding bone (Fig. 5-2). The presence of fibrosis can be assessed on biopsy with specific stains for reticulin and collagen. In normal bone marrow, the immature blast cells are frequently located near the endosteal surface. In MDS, these cells may be distant to this site and form aberrant clusters referred to as abnormal localization of immature precursors (ALIP). Immunohistochemical staining of biopsies can aid diagnosis, with CD34 staining to identify blast and progenitor cells and CD42 or CD62 for quantitation and assessment of megakaryocytes (34) (see Fig. 5-2).
FIGURE 5-2
Morphological and immunohistochemical feature of bone marrow biopsy in the myelodysplastic syndromes. A. Trephine bone marrow biopsy with numerous dysplastic monolobated megakaryocytes in a 60-year-old female patient with refractory anemia with excess blasts type 1. B. CD61 immunohistochemical stain highlighting many dysplastic micromegakaryocytes. CD61 may be helpful in detecting dysplastic micromegakaryocytes to aid in confirming dysmegakaropoiesis and abnormal translocation of megakaryocytes to endosteal surfaces. C. CD34 immunohistochemical staining highlighting the presence of an increased number of blasts and increased blood vessels.
Nonclonal diseases may cause dysplastic morphological changes in blood cells. Secondary causes of dysplasia should be excluded in the initial assessment and can potentially complicate the diagnosis. Blood cell dysplasia is seen with exposure to heavy metals or antituberculous therapies, B12 and folate deficiency, HIV infection, excessive alcohol consumption (33), and occasionally normal aging (35). Dysplastic features are commonly observed after chemotherapy or with the therapeutic use of granulocyte colony-stimulating factor (G-CSF). These diagnoses should be assessed in the history and may require exclusion with further laboratory testing. Diagnostic difficulties may occur in patients with marked hypocellularity of the bone marrow and in patients with prominent fibrosis as there are often very few cells in the aspirate sample to allow morphological assessment of dysplasia. For patients with prominent hypocellularity of the marrow, it may be difficult to distinguish from aplastic anemia, for which morphological dysplasia of the erythroid lineage may also be observed. In cases of marked fibrosis, bone marrow aspiration is often unsuccessful. Some patients with mild dysplastic changes in the bone marrow and a diploid karyotype may be difficult to definitively diagnose at initial presentation and may require a period of observation to confirm the underlying diagnosis. These patients require review with repeat investigations performed in 3 to 6 months.
CYTOGENETIC AND MOLECULAR ANALYSIS
A cytogenetic abnormality is found in 40% to 50% of patients with primary MDS and lower risk disease; it is higher in patients with more advanced MDS. Cytogenetic analysis of hematopoietic cells derived from the bone marrow aspirate provides important prognostic and predictive information and may direct therapy (eg, lenalidomide therapy with deletion 5q or earlier alloSCT with complex or adverse karyotypes). A karyotypic abnormality provides evidence for the presence of a clonal blood disorder, which may be particularly important if the morphological changes are not clear. Typically, cytogenetic analysis will assess 20 bone marrow metaphases (34). Multiple different cytogenetic abnormalities have been described (36). These are summarized in Table 5-2. No specific cytogenetic abnormalities characterize MDS. Unlike AML and chronic myeloid leukemia, genetic translocations are rare in MDS, but deletions are common.
| Karyotype, No. (%) | |||||||
|---|---|---|---|---|---|---|---|
| Classification | No. | Normal | del(5q) | –7/del(7q) | +8 | –20/del(20q) | Complex |
| All FAB | 1949 | 942 (48.3) | 295 (15.1) | 209 (10.7) | 162 (8.3) | 86 (4.4) | 282 (14.5) |
| RA | 573 | 267 (46.6) | 139 (24.3) | 30 (5.2) | 37 (6.5) | 31 (5.4) | 47 (8.2) |
| RARS | 252 | 147 (58.3) | 23 (9.1) | 24 (9.5) | 14 (5.6) | 9 (3.6) | 20 (7.9) |
| RAEB | 415 | 179 (43.1) | 71 (17.1) | 60 (23.8) | 39 (9.4) | 21 (5.1) | 98 (23.6) |
| RAEB-t | 305 | 132 (43.3) | 38 (12.5) | 50 (16.4) | 30 (9.8) | 16 (5.2) | 68 (22.3) |
| CMML | 272 | 170 (62.5) | 4 (1.5) | 23 (8.5) | 18 (6.6) | 2 (<1) | 12 (4.4) |
| MDS-AL | 132 | 47 (30.9) | 20 (15.2) | 22 (16.7) | 25 (18.9) | 7 (5.3) | 37 (28.0) |
| All WHO | 595 | 285 (47.8) | 110 (18.5) | 53 (8.9) | 40 (6.7) | 22 (3.7) | 71 (11.9) |
| 5q- syndrome | 61 | 0 (0.0) | 61 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| RA | 56 | 38 (67.9) | 3 (6.5) | 5 (10.9) | 1 (2.2) | 1 (2.2) | 6 (13.0) |
| RARS | 26 | 23 (88.5) | 0 (0.0) | 0 (0.0) | 1 (3.8) | 0 (0.0) | 0 (0.0) |
| RCMD | 164 | 88 (53.7) | 11 (6.7) | 20 (12.2) | 12 (7.3) | 8 (4.8) | 18 (11.0) |
| RSCMD | 77 | 34 (44.2) | 8 (10.4) | 8 (10.4) | 8 (10.4) | 3 (3.9) | 12 (15.6) |
| RAEB-I | 90 | 42 (45.7) | 16 (17.8) | 10 (11.1) | 5 (5.6) | 4 (4.4) | 15 (16.7) |
| RAEB-II | 121 | 60 (49.6) | 11 (9.1) | 8 (6.6) | 13 (10.7) | 5 (4.1) | 19 (15.7) |
The presence or absence of a cytogenetic abnormality has a marked influence on prognosis (36). Median survival of patients with normal karyotypes is approximately 53 months, compared to less than 12 months for patients with three or more cytogenetic abnormalities (complex). Del(5q) and del(20q) are associated with a favorable prognosis. However, when these abnormalities are present in association with other cytogenetic abnormalities, especially as a component of a complex karyotype, the prognosis is poor. Abnormalities of chromosome 7, usually deletions, are associated with poor prognosis regardless of the presence or absence of other abnormalities. Complex cytogenetic abnormalities are more frequently observed in patients with increased marrow blasts. A progressively worse prognosis is observed with increasing complexity. Patients with six or more abnormalities have a very poor median survival of 5 months (36). In 2012, a new comprehensive scoring cytogenetic system was developed using data from 2,902 patients (37). This analysis resulted in 19 new cytogenetic categories and 5 prognostic subgroups (Fig. 5-3). This scoring system serves as the basis for the IPSS-R (5).
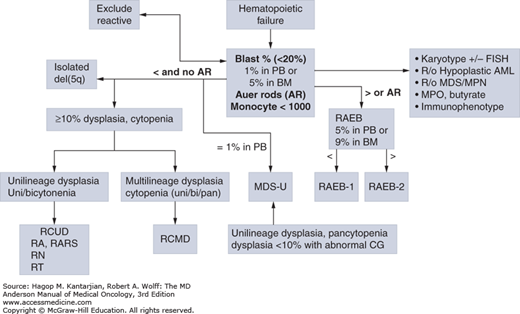
FIGURE 5-3
Algorithm for the classification of adult-onset primary myelodysplastic syndromes (MDS). This classification system is based on the 2008 criteria of the World Health Organization. AML, acute myeloid leukemia; FISH, fluorescence in situ hybridization; RAEB, refractory anemia with excess blasts; RARS, refractory anemia with ring sideroblasts that are equal to or greater than 15% of bone marrow erythroid precursors; RCMD, refractory cytopenia with multilineage dysplasia; RCUD, refractory cytopenia with unilineage dysplasia.
Treatment-related MDS has a particularly high incidence of cytogenetic abnormalities, with karyotypic changes observed in 70% to 90% of cases (26,29,36). A high incidence of abnormalities is associated with an unfavorable prognosis for this group of patients. Abnormalities of chromosomes 5 and 7 are frequently observed after exposure to alkylating agents (38,39). Translocations involving 11q23 are seen after treatment with topoisomerase II inhibitors (38).
The high frequency of chromosomal deletions has prompted interest in the identification of epigenetic repressive alterations, such as aberrant DNA methylation in MDS. Aberrant DNA methylation of multiple promoter CpG islands has been associated with poor prognosis in MDS (40). At this point, we do not have evidence of specific molecular pathways that are epigenetically inactivated in MDS.
An association between a genetic abnormality and disease phenotype was reported in a few specific subsets of MDS. An example is the 5q- syndrome. A minority of patients with an interstitial deletion of chromosome 5q display an indolent anemia with relative preservation of the platelet count associated with hypolobated megakaryocytes in the bone marrow. This array of findings is called the 5q- syndrome (41) and is recognized as a separate diagnostic entity in the current WHO classification. The genetic defect within the deleted region that is responsible for the disease is not known. Research has focused on one candidate gene, SPARC, that may potentially contribute to the malignant phenotype (42). CTNNA1 is another gene on chromosome 5q identified to be important in MDS and AML but without specific features of the 5q- syndrome (43). Ebert et al. reported the identification of RPS14 as haploinsufficient in 5q- MDS. RPS14 is involved in ribosomal biogenesis, and its deficiency has a role in anemia in this syndrome (44). It is likely that a complex network of genes cooperate in the pathogenesis of this syndrome. Indeed, microRNA 145 and 146a have been found to be involved in the biology of 5q- syndrome (45).
A small number of patients have been described with a deletion of 17p associated with abnormalities in the p53 gene. This specific disorder has a poor prognosis and may be suspected when morphological characteristics of prominent dysgranulopoiesis, including neutrophils exhibiting the Pelger-Huet anomaly and abnormal vacuolization, are present (46). Acquired hemoglobin H disease produces red cell changes on the blood smear reminiscent of α-thalassemia. This red cell phenotype is secondary to decreased expression of α-globin within the bone marrow MDS clone and is associated with a mutation in the ATRX gene in most cases (47). These rare syndromes represent a small minority of patients with MDS, and specific gene defects are not identified in most patients.
Several groups have used large-scale single-nucleotide polymorphism (SNP) arrays in MDS (48). This has allowed the identification of areas of microdeletions and uniparenteral disomy in MDS (49). It is likely that these genomic regions harbor genes important in MDS, such as c-CBL (50). Over the last 5 years, we have accumulated significant information regarding genomic alterations in MDS (30,51). The data are summarized in Fig. 5-4 from a study reported by a consortium of European investigators (51). Common mutations affect genes involved in gene splicing, epigenetic regulators (eg, TET2, DNMT3A, ASXL1, and EZH2), and other pathways, such as TP53. The presence of mutations in TP53 and EZH2 is associated with poor prognosis (30). A recent analysis correlated specific genomic alterations with gene expression patterns that may explain some of the phenotypic features of the disease (52).
OTHER LABORATORY STUDIES
Flow cytometry is not required for the routine diagnosis of MDS, but it may sometimes provide valuable supplementary information. Flow cytometry can confirm the presence of specific myeloid lineages within the marrow and may also identify aberrant expression of cell surface markers indicative of a clonal cell population. This may have diagnostic significance in confirming abnormal hematopoiesis, particularly in the setting of inconclusive morphological changes and a normal karyotype. Quantitation of the number of CD34-positive cells in the bone marrow may assist in the differentiation of hypoplastic MDS from aplastic anemia. In MDS, the number of CD34 cells is usually normal or increased, compared to aplastic anemia, for which it is frequently reduced (53).
Fluorescent in situ hybridization (FISH) techniques using probes specific for specific chromosomes (ie, covering chromosomes 5, 7, 8, 20) have not been fully standardized in MDS. Their use should not be consider standard of care in MDS and cannot yet replace conventional cytogenetics.
DIAGNOSIS
The classification systems used to group different MDS have evolved over time with increased understanding of the biology and genetics of the disease. The first widely accepted classification was that proposed by the French-American-British (FAB) study group (54). The FAB categorized MDS primarily on the percentage of blasts in the peripheral blood and bone marrow, with disease entities defined by increased numbers of blasts associated with a more aggressive clinical course. Patients with a bone marrow blast percentage greater than 30% were considered to have acute myeloid leukemia. This classification used only morphological criteria to define disease groups and provided a framework that allowed the study of the natural history of MDS and its response to therapy.
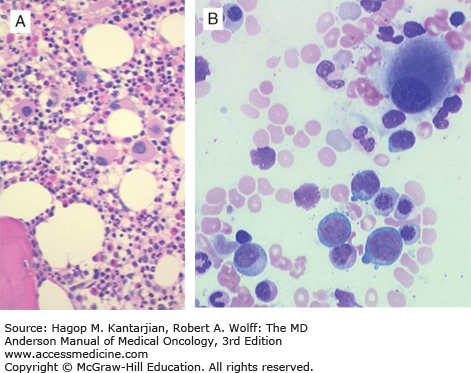
The WHO classification of MDS was developed with the objective of using all features of disease biology, including morphology, cytogenetics, immunophenotype, and clinical behavior (55). This classification was updated in 2008 (56) (see Table 5-1). Figure 5-5 shows an algorithm for the morphological diagnosis of MDS. In the original WHO classification, the importance of morphological assessment of blast percentage within the bone marrow and peripheral blood was retained, although the threshold level for the diagnosis of acute leukemia was altered. Patients with more than 20% bone marrow blasts were considered to have AML. Patients with 20% to 29% blasts had a similar prognosis as patients with greater than 30% blasts. Within the WHO MDS categories with an increased blast percentage, the magnitude of the blast elevation was quantified between refractory anemia with excess blasts (RAEB) types 1 and 2, reflecting the worse prognosis of patients with an elevated blast count (2). In patients with a normal proportion of blast cells within the bone marrow, the relatively indolent refractory anemia (RA) and RA with ring sideroblasts (RARS) introduced in the FAB system were further delineated by assessment for the presence of multilineage dysplasia. Patients with dysplastic maturation limited to the erythroid lineage have a more favorable prognosis than patients with cytopenia and dysplasia present in multiple myeloid lineages. Also, WHO introduced the 5q- syndrome as a separate diagnostic entity primarily on the basis of a genetic abnormality rather than morphological features alone. Deletions involving chromosome 5q are relatively common in MDS, and the WHO classification tightly defined the syndrome as an isolated del(5q) associated with anemia, a preserved or increased platelet count, and hypolobated megakaryocytes on the bone marrow biopsy (Fig. 5-6). The WHO classification has been validated by a number of independent groups (57,58). The most recent WHO classification (56) included the following changes: (1) specific guidelines for the requirement of specimen collection and blast and blast lineage assessment, as well as for the analysis of genetic alterations; (2) an effort to report new changes in the diagnosis and classification of MDS/MPN; (3) inclusion of patients with cytopenias but not clear morphological evidence of MDS in the bone marrow as presumptive MDS; (4) inclusion of refractory cytopenia with unilineage dysplasia; and (5) disappearance of the category of refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS) (56).
FIGURE 5-5
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


