Ref.
n
BMI (kg/m2)
Testosterone ng/dL
(nmol/L)
SHBG nmo/L
MetS
HOMA-IR
Pre
Post
Pre
Post
Pre
Post
Pre
Post
Pre
Post
[148]
22
46.2 ± 0.9
−16.6%
340 ± 21
636 ± 186
+21.6
[149]
33
50.3 ± 6.1
31.5 ± 4.7
248 ± 91
546 ± 165
18.3 ± 11.8
42.7 ± 18.1
[26]
20
43.6
34.8
8.1
13.2
19
39
[150]
46.9 ± 7.8
30.9 ± 5.0
256 ± 121
508 ± 161
25.0 ± 12.3
48.5 ± 16.1
11.8 ± 7.8
3.5 ± 4.7
[151]
20
50.4 ± 8.7
302 ± 102
[152]
28
50.1 ± 11.2
35.9 ± 7.02
(8.31 ± 3.24)
(12.7 ± 3.8)
23.4 ± 17.4
37.8 ± 18.5
75%
25%
Overall, why SHBG is associated with the MetS and to a lesser extent with T2DM, and the role SHBG plays in determining how androgens or estrogens influence MetS and T2DM risk are only partly understood, and may differ by sex, as discussed below.
SHBG: Regulator of Androgen and Estrogen Action
Plasma SHBG is produced as a glycoprotein by hepatocytes, and is released into the blood circulation as a homodimer of identical subunits, each of which contains a single steroid-binding site for active androgens and estrogens [12]. Human SHBG binds 5α-dihydrotestosterone (DHT) with about 5 times greater affinity than testosterone and about 20 times higher affinity than estradiol [28]. There are sex differences in human SHBG production by the liver at different stages of the life cycle, and the occupancy of SHBG steroid-binding sites in men and women differs greatly [11]. Plasma levels of SHBG are usually about twofold higher in women than in men but far fewer SHBG steroid-binding sites are occupied in women than in men, in whom SHBG is largely occupied by testosterone [11, 28]. The obvious corollary of this sex difference is that SHBG functions differently in women than in men.
In women, SHBG appears to play a primary role in limiting the access of both testosterone and estradiol to their target cells. This has been evident ever since the early findings that low plasma SHBG levels are directly associated with increased bioavailable testosterone and symptoms of hyperandrogenism [29]. To illustrate this further, marked 5–10 fold increases in plasma SHBG levels after administration of some oral contraceptives accentuate reductions in free testosterone levels, and are often used to reduce hyperandrogenic symptoms, including hirsutism [30]. In addition, low plasma SHBG levels in obese postmenopausal women are linked to increase risk for endometrial cancer because they increase free estradiol levels [31]. Plasma levels of SHBG also increase by 5–10 fold in women during pregnancy, and it is assumed that this is caused by estrogen-dependent increases in liver SHBG production [29]. The functional importance of elevated plasma SHBG levels during pregnancy is unclear but it may limit the exposure of pregnant women to fetal adrenal androgens that escape metabolism by the placenta, as illustrated by the transient manifestations of severe androgen excess in a pregnant woman with a rare case of SHBG deficiency due to mutations with both SHBG alleles [32]. Women with gestational diabetes have lower plasma SHBG levels [33], and recent reports indicate that low pre-pregnancy levels of SHBG are associated with increased risk for gestational diabetes even in women who were of normal weight with no previous history of this condition [34].
Measurements of plasma SHBG and total testosterone levels are used together to calculate the plasma concentrations of free testosterone, which are generally considered to be the most reliable measure of androgenic activity in both sexes [35]. However, the mathematical algorithms used for this purpose are less than optimal, and the recent identification of human SHBG variants with abnormal steroid-binding activities or immunological properties highlights the pressing need for more direct methods to measure free testosterone levels based on sensitive and specific mass spectrometric methods [36]. The following sections summarize recent advances in our understanding of the functional properties of SHBG, as well as its production by the liver in health and disease, and highlight gaps in our knowledge that remain to be filled.
Structure and Function of Human SHBG
The evolutionary origins of SHBG remain obscure, but its monomeric form comprises two laminin G-like (LG) domains found in the C-terminal regions of several extracellular proteins (Fig. 15.1a), including protein S and gas6 [37–39]. Protein S and gas6 are potential ligands for the Axl/Sky subfamily of receptor tyrosine kinases and they interact with these receptors through their “SHBG-like domains” [40]. Protein S and gas6 do not bind steroids, and SHBG appears to have acquired its function as a steroid-binding protein early in the evolution of vertebrates because it is present in the fishes [41, 42].
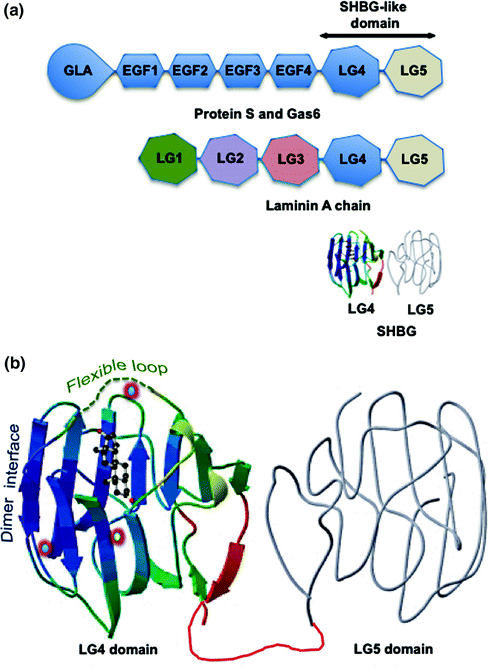
Fig. 15.1
Human SHBG and its structural and functional properties. a Domain structures of human SHBG and other proteins with tandem Laminin G-like (LG) domains. Human SHBG resembles the LG4 and LG5 domain structure found in the C-terminus of the laminin A chain. Protein S and Gas6 are large extracellular proteins that comprise a GLA (multiple γ-carboxyglutamic acid) domain followed by several EGF-like domains and similar tandem LG4 and LG5 “SHBG-like domain” that interacts with the Axl/Sky subfamily of receptor tyrosine kinases. b The crystal structure of the SHBG LG4 domain in complex with 5α-dihydrotestosterone (DHT) together with the predicted structure of the LG4 domain (gray). The unstructured flexible loop region that covers an entrance to the steroid-binding pocket is shown as a dashed green line, and the dimer interface that holds two monomers together in a head to head manner to form the SHBG homodimer is also indicated. The position of the calcium atom (yellow center), the binding of which is essential for maintaining dimerization and the integrety of the steroid-binding is shown, as are the positions of the two zinc atoms (blue center); including the zinc atom that is held in position by a histidine in the flexible loop region of human SHBG and influences the specificity of steroid binding. The DHT is positioned in the steroid-binding site
The quaternary structure of the SHBG homodimer has never been resolved but there is evidence that it adopts a torpedo-like head to head conformation [43]. The N-terminal LG domain contains the steroid-binding site and the dimerization domain (Fig. 15.1b), and its crystal structure has been determined with several different androgens or estrogens in the steroid-binding site [44–46]. These structures show how different steroids are positioned within a hydrophobic binding pocket, and how critical amino acid residues participate in the steroid-binding interactions. In particular, they indicate that androgens and estrogens are accommodated very differently within the hydrophobic core of each SHBG monomer, and that the binding of androgens and estrogens exert differences in the positioning of specific residues on their surface [46], including those within a flexible loop region [47] that covers an entrance to the steroid-binding pocket (Fig. 15.1b). The positioning of this flexible loop has been proposed to influence the flux of androgens and estrogens into and out of the binding site differently, but an additional entrance or exit to the steroid-binding site for estrogens may also exist that is not accessible to androgens [36].
High-resolution crystal structures have also revealed the mechanism of how the SHBG monomers associate as a homodimer, and show that both monomers of the homodimer can bind a steroid ligand [48]. What remains to be determined is whether occupancy of the steroid-binding site of one monomer exerts allosteric effects that influence the steroid-binding affinity of the partner monomer, and whether androgens and estrogens exert different effects in this regard. If this is the case, it may have a different impact of the function SHBG in women in whom steroid occupancy of the binding sites is less than 20%, while in men ~50% of the sites are normally occupied by androgens [11, 28].
It has also been known for many years that calcium maintains the integrity of the SHBG homodimer and its ability to bind steroids with high affinity [49]. This explains why the steroid-binding activity of SHBG in EDTA plasma is disrupted by freeze thaw cycles. Structural resolution of the N-terminal LG domain of SHBG identified the calcium-binding site responsible for this (Fig. 15.1b), and provided evidence that calcium binding exerts long-range allosteric effects that integrate both dimerization and steroid-binding activities [44–46]. The crystal structures also revealed the presence of two zinc-binding sites in the N-terminal LG domain (Fig. 15.1b); one of which is highly conserved across species and is probably structurally important, although this has not been formally proven [50]. The other zinc-binding site is located within the loop region that covers the steroid-binding site, and a zinc atom is held in place by a histidine residue in this location (Fig. 15.1b). Importantly, occupancy of this zinc-binding site specifically reduces the binding affinity of human SHBG for estrogens but not androgens [50]. While this may not be relevant to the activities of SHBG in blood where free zinc concentrations are very low, it may be relevant in other extravascular tissue compartments where they are particularly high, such as in the prostate and male reproductive tract.
Although the SHBG C-terminal LG domain structure has not been resolved, it can be modeled (Fig. 15.1b) on that of the N-terminal LG domain [12]. The C-terminal LG domain does not participate in steroid binding or dimerization, but it normally contains two sites for N-linked glycosylation. One of these N-glycosylation sites is invariably conserved across vertebrate species and is predicted to be functionally important [51]. There is also a relatively common human SHBG variant (SHBG D327 N) that contains an additional N-glycosylation site within the C-terminal LG domain [52].
Well before the structure of SHBG was resolved, reports appeared describing interactions between SHBG and other proteins, especially those within the plasma membranes of cells [53, 54], leading to proposals that the function of SHBG extends beyond those of a plasma transport protein [55]. These functions included cell-signaling activities that may explain in part the non-genomic actions of steroids [56] or the endocytotic delivery of steroids to their target cells [57]. The realization that SHBG was structurally related to the proposed receptor-binding domains of protein S and Gas6 [40] fueled interest in these ideas, as did the demonstration that the nonspecific endocytotic receptor megalin is capable of recognizing SHBG and internalizing it together with its ligand [58]. However, functional receptors for SHBG that exert activities in physiological contexts remain to be identified. The most likely reason for this is that early observations of interactions between SHBG and cell membrane proteins were made using purified preparations of SHBG and their potential targets, or tissue culture systems where purified SHBG was added in the absence of other plasma proteins. These experimental designs neglected the fact that SHBG is a trace protein in biological fluids, and there are many other proteins including those that have structurally related domains that could compete with its binding to membrane proteins in vivo.
However, an interaction between SHBG and the extracellular matrix-associated proteins, fibulin-1D and fibulin-2 has been identified under conditions that satisfy these experimental caveats [59]. These members of the fibulin family not only recognize and sequester SHBG from dilutions of plasma samples that mimic those in extravascular tissue compartment, but do so in a ligand-dependent and ligand-specific manner [59]. Moreover, the interaction was favored when the SHBG steroid-binding site was occupied by estradiol in vitro as well as in vivo in the endometrial stroma [59]. In mice expressing a human SHBG transgene, SHBG also leaves the blood vasculature in the endometrial stroma and accumulates in the stromal matrix when estrogens levels are elevated during the estrus cycle [59]. These observations at least provide direct evidence that SHBG is capable of acting in extravascular compartments of some tissues to modulate the accessibility of sex steroids to their target cells.
Tissue Specific Expression of Human SHBG
The SHBG locus is located within the short arm of human chromosome 17 [60], and comprises multiple transcription units defined by the utilization of different promoter regions that flank alternative exon 1 sequences [61–63]. These studies indicated that SHBG encodes a large number of transcripts, which all contain alternative exon 1 sequences spliced to a common exon 2 sequence followed by up to six other exon sequences, but only two of them have been shown to encode a functional SHBG protein (Fig. 15.2). The biologically most important of these is the mRNA in the liver that encodes the SHBG precursor polypeptide that includes the secretion signal polypeptide sequence encoded by exon 1 within an ~4.5 kb SHBG transcription unit [12]. By contrast, the major SHBG transcript in the testis [61] contains a noncoding alternative exon 1 sequence (Fig. 15.2), and there is evidence that an AUG translation initiation codon within exon 2 produces an N-terminally truncated SHBG isoform starting with Met30 in the mature SHBG sequence [64]. This SHBG isoform is produced in male germ cells and is not secreted, but it appears to bind steroids in same way as plasma SHBG and accumulates in the acrosome of sperm [64]. Although the function of this human sperm SHBG isoform is unknown, its concentration in the acrosome declines with age and correlates with sperm motility [65].
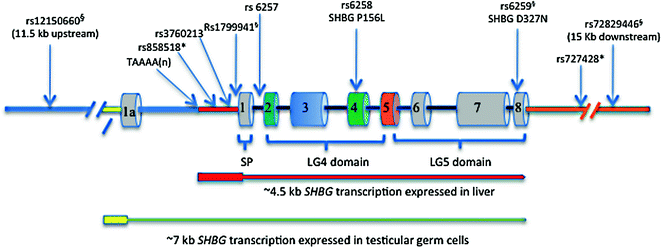
Fig. 15.2
Structural organization of the human SHBG locus, showing the two main transcription units expressed in the liver and male germ cells, together with the positions of common polymorphism associated with individual differences in the production or function of plasma SHBG. The ~4.5 kb transcription unit for the SHBG mRNA in the liver that encodes the SHBG precursor polypeptide (encoded by exons 1–8) includes ~800 bp of promoter sequence upstream of exon 1 that encode the secretion polypeptide (SP) that is remove co-translationally to yield the mature SHBG monomer of LG4 and LG5 domains. The ~7 kb transcription unit encodes the most abundant alternative SHBG transcript found in testicular germ cells, and it comprises an alternative exon 1a sequence spliced into exons 2–8. Exon 1a is noncoding but translation can initiate from the AUG codon (in exon 2) for Met30 in the mature SHBG polypeptide to yield an N-terminally truncated SHBG isoform that accumulates in the acrosome. Those SNPs in linkage disequilibrium with each other are indicated with symbols (* or §), respectively
Cell-specific expression of SHBG in the human testis differs from that observed in subprimate species in which the gene is expressed in Sertoli cells resulting in the production of the SHBG homologue, known as the androgen-binding protein, which is secreted into the seminiferous tubules where it is thought to act to promote sperm maturation within the male reproductive tract [66]. Although SHBG is present in human seminal plasma [67], it does not originate from Sertoli cells because the human SHBG promoter contains a binding site for USF transcription factors that repress transcription in Sertoli cells, and these sequences are absent the SHBG genes of subprimate species [64]. This species difference in the cell type expression of SHBG in the testis is enigmatic but may be related to differences in the spatial organization of spermatogenic cells within the seminiferous tubules.
None of the other alternative exon 1 containing SHBG transcripts encodes a secretion signal polypeptide in frame with coding sequences for an SHBG-like polypeptide. However, “humanized mice” expressing an SHBG transgene have demonstrated that extrahepatic expression of SHBG occurs in some cell types, such as the androgen sensitive epithelial cells of the proximal convoluted tubules of the kidney, and much of the human SHBG produced by these cells remains within them [68]. Furthermore, the SHBG that accumulates within these cells is N-terminally truncated and incompletely glycosylated, much like the SHBG isoform in the acrosome [65], and it appears to act to sequester DHT from the extracellular environment and modify its access to the androgen receptor [69]. This raises the possibility that the alternative SHBG transcripts identified in human prostate cancer cells [62, 63] encode SHBG isoforms that function within the cells rather than having to be secreted to act, but this will only be resolved when these intracellular SHBG isoforms are actually isolated and characterized from human tissues.
Regulation of Plasma SHBG Production by the Liver
Early studies indicated that SHBG production by the liver is under hormonal, nutritional and metabolic control [29]. Reductions in plasma SHBG in boys during puberty were initially thought to reflect a negative effect of androgen on SHBG production, but this was difficult to reconcile with a lack of any increase in plasma SHBG levels in men after orchiectomy [70]. A normal pubertal decline in SHBG was also been reported in siblings with complete androgen insensitivity, implying that the substantial reductions in SHBG levels in boys during puberty are not androgen-dependent [71]. Importantly, these changes in SHBG levels during puberty in boys occur with advancing age irrespective of any changes in total testosterone [72], and cause increases in albumin-bound and free testosterone that likely contributes to male sexual development [73]. Clearly other factors account for reductions in plasma SHBG levels in girls during puberty, and there is evidence that increases in body weight and composition are the main operative agents of these changes [74]. Reductions in plasma SHBG are more pronounced in girls with premature puberty, many of whom are overweight, and SHBG measurements have be useful as a prognostic indicator of risk for T2MD in these individuals [75], as highlighted in a recent review [76].
In healthy adults, SHBG levels remain relatively constant, and in elderly men and women changes in plasma SHBG are modest, with modest age-related increases in men during old age that has been attributed to GH deficiency [77]. Small reductions in SHBG levels in postmenopausal women are unlikely to be related to a loss of ovarian estrogen because transdermal hormone replace therapy does not influence plasma SHBG levels [78], and are more likely to associated with changes in body composition [79], hormones like leptin and adiponectin [80], thyroid hormone status [81], or the metabolic state of the liver [82].
Early reports that plasma SHBG levels are very low in obese men [83], but very high in patients with anorexia nervosa [84], provided the first indication that nutritional status or metabolic factors influence the production or plasma clearance of SHBG. It was also recognized early on that plasma SHBG levels are elevated in hyperthyroid individuals and in patients after treatment with thyroid hormone [29], which itself exerts profound effects on metabolic state.
The very low levels of SHBG in obese men and women remained unexplained until it was reported that treatment of human HepG2 hepatoblastoma cells with insulin decreased SHBG production in vitro [85]. This provided credence to the idea that elevated insulin levels were responsible for the low plasma SHBG levels in insulin-resistant obese individuals, but the cellular mechanisms responsible for this have never been identified. Although, SHBG levels are normal in children with type 1 diabetes [86], they are in the upper normal ranges in men and women with type 1 diabetes, and display an inverse correlation with fasting insulin levels [87]. While the latter observations were interpreted as confirmation of a negative effect of insulin on hepatic SHBG production, a strong association between SHBG and glucose levels was also observed in subjects with T2DM in the same study.
Hyperglycemia is the primary consequence of deficiencies in insulin production or action in patients with type 1 diabetes or T2DM, and the question of how nutrition and metabolic state might influence plasma SHBG production by the liver was therefore re-examined in HepG2 cells, as well as in humanized mice that express human SHBG transgenes in the liver under the control of the SHBG promoter [88]. These studies failed to confirm a direct effect of insulin on SHBG expression in the liver in vitro or in vivo. Instead they demonstrated that SHBG is regulated in the liver by the monosaccharides, glucose and fructose, rather than insulin. Moreover, they presented a detailed mechanism for how these effects are mediated at the level of SHBG transcription [88], thereby providing the first molecular explanation for why plasma SHBG levels are so low in patients with the MetS [4, 89]. The finding that monosaccharides, and fructose in particular, potently downregulated SHBG expression in hepatocytes also provided an explanation for why the wide-spread use of high fructose corn starch as a food and beverage supplement has been associated with the remarkable increases in the prevalence of the MetS and T2DM worldwide.
The transcription unit responsible for SHBG production by the liver was originally identified through experiments using different human SHBG transgenes in mice [68], as well as with SHBG promoter-driven reporter constructs in HepG2 cells [90]. Together these studies demonstrated that the ~4.5 kb transcription unit, which includes an ~800 bp promoter flanking exon 1 (Fig. 15.2), is sufficient to drive SHBG transcription in the liver. The use of DNase I footprinting assays and reporter gene assays also allowed the mapping of cis-elements within the promoter that interact with hepatic nuclear protein extracts [90]. The latter studies revealed that the binding of HNF4α to a nuclear hormone receptor (NHR) response element close to the SHBG transcription start site compensates for lack of a TATA box, and substitutes for the TATA-binding protein in recruiting the transcriptional machinery to the proximal promoter of SHBG. At that time, the realization that HNF4α plays a key role in the regulation of SHBG expression was particularly enlightening because HNF4α had just been identified as the first gene to be implicated in Maturity Onset Diabetes of the Young [91].
As a member of the NHR family of transcription factors, HNF4α, binds to palindromic DNA response elements that also bind other members of the NHR family, including the COUP-TFs [92]. Importantly, HNF4α and COUP-TF1 compete for the same response element in the SHBG promoter and to exert opposing actions on SHBG transcription, with HNF4α increasing transcription while COUP-TF1 represses it [90]. This explained for the first time how HNF4α and COUP-TF1 might serve as the on-off switches of SHBG transcription in hepatocytes under a wide range of pathophysiological conditions related to the MetS.
The underlying mechanism of how oral glucose and fructose downregulate hepatic SHBG production involves their rapid hepatic metabolism into acetyl-CoA, the immediate precursor of palmitic acid: the first of the long chain fatty acids produced in the liver [88]. A buildup of palmitate in HepG2 cells after treatment with monosaccharides is indicative of increased lipogenesis, and occurs in concert with reductions in HNF4α and SHBG mRNA levels, and concurrent reductions in SHBG secretion [88]. A strong positive correlation between hepatic HNF4α and SHBG mRNA levels has been also observed in a study of male and female cancer patients [93]. Furthermore, in the later study, hepatic HNF4α and SHBG mRNA levels were also inversely related to hepatic triglyceride levels and decreased in relation to body mass index. Why HNF4α levels decrease in hepatocytes under conditions where lipogenesis is enhanced may be related to the fact that fatty acyl Coenzyme A thioesters and palmitoleic (C16:1) and oleic acid (C18:1) were originally identified as potential ligands of HNF4α [94–96], which may induce conformational changes in HNF4α [97] and somehow destabilize it. It is also possible that other dietary fatty acids, such as linoleic acid, and long-chain fatty acid metabolites bind HNF4α and alter its levels or activity as a transcription factor [98]. This of interest because recent studies have indicated that diets rich in linoleic acid may increase plasma SHBG levels [99].
This interplay between HNF4α and COUP-TF as key regulators of SHBG expression in hepatocytes is emerging as a common mechanism that accounts for other links between abnormal plasma SHBG levels and clinical conditions related to an abnormal metabolic state. In patients with the MetS, an increase in free fatty acid flux due to a failure to suppress lipolysis in adipocytes will increase hepatic free fatty acids [100], and thereby contribute to reductions in hepatic HNF4α levels and SHBG expression. By contrast, high plasma levels of SHBG are usually found in individuals with hyperthyroidism, while low levels are associated with thyroid hormone deficiencies [29] and the reason for this is that thyroid hormone influences hepatic SHBG production indirectly by altering hepatic HNF4α levels [101]. Low plasma SHBG levels are also frequently observed in patients with hepatic steatosis that predisposes to T2DM [102], and have been independently associated with nonalcoholic fatty liver disease (NAFLD) in subjects with T2DM [103]. The low plasma SHBG levels in NAFLD patients likely reflects the substantial accumulation of fatty acids in their livers [104], including palmitic acid that is linked to reduced hepatic HNF4α levels [88].
Increases in the plasma levels of pro-inflammatory cytokines (TNFα and IL-1β) and reduced levels of adiponectin, which are typically seen in overweight individuals at risk for the MetS [105], have been recently shown to downregulate [106, 107] and upregulate [108] SHBG production by the liver, respectively, and they also do this by changing hepatic HNF4α levels. By contrast, the transcription factor PPARγ interacts with a different NHR response element in the SHBG proximal promoter to repress transcription [109], and this probably accounts for the lower plasma SHBG levels in subjects with the hyperactive 12 Ala PPARγ variant [110] that has been linked to low risk for T2DM and myocardial infarction [111]. However, the use of insulin sensitizers, like pioglitazone, that act by stimulating PPARγ, increase plasma SHBG levels in individuals with the MetS [112]. This may be explained by the fact that the NHR response element in the SHBG proximal promoter that binds PPARγ can also bind HNF4α [90], as well as several other orphan members of the NHR family, including CAR and LXR, which can all potentially compete for binding at this site. Improvements in hepatic metabolic state induced by pioglitazone and other insulin sensitizers, likely change the hepatic complement of these orphan NHRs, which may account for changes in SHBG expression after their use.
Lessons from Naturally Occurring SHBG Polymorphism
Individual differences in plasma sex steroid and SHBG levels were recognized as an inherited trait in early studies [113], but the genetic basis for this was not appreciated until human SHBG was cloned and sequenced [61, 114]. The first SHBG variant to be characterized at the genetic level explained why SHBG monomers in the serum of some individuals migrate with an unusually large molecular size in denaturing polyacrylamide gels [38, 115], and was shown to be caused by an Asp327 to Asn substitution that introduces an additional consensus site for N-glycosylation and an extra N-liked oligosaccharide per monomer [52]. The nonsynonymous SNP (rs6259) responsible for the SHBG D327 N variant (Fig. 15.2) is common in most ethnic groups globally. While the steroid-binding activity of SHBG D327 N is normal [52], a reduction in its plasma clearance rate may account for why carriers of SHBG D327 N have slightly higher plasma SHBG levels [116], and in part explain reports that SHBG D327 N has a protective effect on T2DM risk [117, 118].
Several SHBG polymorphisms linked to differences in plasma SHBG levels are located in noncoding regions of the gene (Fig. 15.2). The first of these to be identified was the TAAAA(n) polymorphic repeat that is part of an Alu sequence positioned about 1 kb upstream from the transcription start site for the SHBG mRNA [119]. Variations in number of this pentanucleotide repeat per SHBG allele have been examined in relation to plasma SHBG levels in patients with a range of clinical conditions [120], and the emerging consensus is that men with low numbers of TAAAA repeats have lower SHBG levels are at greater risk for the MetS [121].
The molecular explanation for how the variable TAAAA repeat number influences hepatic SHBG production remains to be fully elucidated, but studies using reporter genes driven by SHBG promoter sequences in HepG2 cells have demonstrated that the pentanucleotide repeat number may influence SHBG transcription through interactions with other downstream elements within the promoter including an SP1 binding site [119]. However, the effect of variable TAAAA repeats on SHBG transcription observed in these in vitro studies does not match what is observed in terms of differences in plasma SHBG levels in patients [122], and this may be due to either the transformed nature of HepG2 cells or the fact that reporter constructs act as naked DNA without assuming the complex chromatin structure that the SHBG promoter normally adopts in vivo. This has precluded further in depth studies of the molecular basis of how variations in the TAAAA repeat number influence SHBG transcription, but the recent introduction of genome editing techniques should allow this question to be addressed more directly.
Other common SNPs within the SHBG promoter or its first intron have been linked to plasma SHBG levels in genome-wide association studies (GWAS) of sex hormone dependent diseases (Fig. 15.2), including those associated with the MetS and T2DM. One of these SNPs (rs1799941) that is linked to individual differences in plasma SHBG levels, in men [123], women [124, 125], and in children and adolescents [126], is located close (within 10 bp) to the SHBG transcription start site, with A and G alleles being associated with higher and lower plasma SHBG levels, respectively [127]. However it has also been noted that rs1799941 is in linkage disequilibrium (LD) with other genetic variations within the SHBG locus [128], including rs6295 and rs72829446 that is located ~15 kb downstream from the coding sequences for SHBG transcripts (Fig. 15.2). The rs1799941 is also in strong LD with rs12150660 located about 11.5 kb further upstream within the SHBG locus (Fig. 15.2), and which is also strongly associated with plasma testosterone concentrations [10].
A common SNP (rs6257) within intron 1 of the SHBG gene (Fig. 15.2) was originally associated with lower plasma SHBG levels and breast cancer risk [129] but has also been reported as a risk allele for T2DM [117]. This is in contrast to rs6295 that produces the SHBG D327 N variant with a prolonged plasma half-life, which is considered to be protective of risk for developing the MetS [104, 117, 118]. Recently, another SNP (rs3760213) linked to higher levels of plasma SHBG has been associated with a decreased risk for the MetS [118], and it may be directly involved in increasing SHBG expression because it is positioned close to a footprinted region (DNase1 footprint 6) ~200 bp upstream (Fig. 15.2) of the SHBG transcription start site [90]. Another SNP (rs727428) that is associated with plasma androgen and SHBG levels is positioned ~1 kb downstream of the most 3’ SHBG exon [129], and is in LD with an SNP (rs858518) within the promoter that appears to promote SHBG expression in the liver (Fig. 15.2).
Apart from the SHBG D327 N variant (rs6295), there is only one other relatively common non-synonymous SNP (rs6258) within the SHBG coding sequence (Fig. 15.2) that appears to have the capacity to influence plasma testosterone levels. This polymorphism encodes SHBG P156L that has a reduced affinity for testosterone, and men with this variant have lower testosterone levels in their blood, presumably because of an increase in the metabolic clearance of testosterone [10]. Several other naturally occurring SHBG variants have been identified recently with abnormalities in production or the ways they bind their sex steroids ligands, but these occur very rarely and their clinical significance is unknown [36]. However, one of them (SHBG G195E) with a secretion defect [36] has an amino acid substitution in the same position as another rare SHBG variant (SHBG G195R) recently identified in a man with no detectable SHBG in blood plasma [130].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


