CARCINOGENICITY OF INDIVIDUAL TREATMENT MODALITIES
Radiation Therapy
The leukemogenic and carcinogenic effects of ionizing radiation have long been recognized in atomic bomb survivors19–25 and individuals exposed to low doses of radiation for diagnostic purposes or therapeutic radiation for benign conditions.26–30 The relation between radiation dose and cancer risk is well described in these low-dose ranges, with an approximately linear increase in risk with increasing radiation dose up to about 5 Gy for solid tumors,22,31,32 and up to 1.5 to 2 Gy for leukemia.10,23,33,34 The traditional view has been that increased cell killing occurs in the higher dose region and, therefore, results in a lowered cancer risk, whereas tissues exposed to intermediate doses are at highest risk for cancer development. A classic radiation dose-response curve plotting the incidence of cancer against the dose of radiation absorbed has a characteristic shape, with the incidence of malignancy increasing with a dose up to a maximum of about 3 to 10 Gy, followed by a decrease in cancer risk with a further increase in dose.34,35
Much of the data on the leukemogenic and carcinogenic effects of radiation in the higher dose range were derived from cancer survivors who have received therapeutic radiation.9,10 In assessing radiation-related SPCs in these patients, it is important to take into consideration other potential contributing factors. These include exposure to cytotoxic drugs (see Chemotherapy section), genetic predisposition,36–40 immunosuppression,41,42 infections,42–44 environmental exposures,45 and heightened surveillance in cancer survivors.46 Additional factors can modify the effect of radiotherapy on SPC risk. Patients with certain cancer prone syndromes, including hereditary retinoblastoma (RB), nevoid basal cell carcinoma syndrome, and neurofibromatosis-1 are more susceptible to radiation-induced cancer.36,47–49 More recently, genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNP) associated with sensitivity to radiation-related cancers.50–52 Tobacco history increases the risk of treatment-related lung cancer in a multiplicative manner as demonstrated in a case-control study of HL survivors.53 Younger age at irradiation is associated with a significantly increased risk of breast cancer54–59 and thyroid cancer,54,60 as shown in survivors of HL, breast cancer, and childhood cancer. On the other hand, early menopause has a protective effect on breast cancer after radiotherapy in HL survivors,61,62 suggesting that ovarian hormones play an important role in promoting tumorigenesis once an initiating event has been induced by radiation.
More recent studies have addressed the effect of radiation dose in the therapeutic range on SPC risk by estimation of radiation dose at the site of SPC development. The risk of radiation-related cancer has been shown to increase with increasing dose for cancers of the breast,59,61–63 esophagus,64 lungs,53,65 stomach,66 meningioma,67 and sarcoma,68–70 with no evidence of a downturn in risk at high doses within the therapeutic range. Preliminary data also show an increasing risk of secondary pancreas cancers with increasing radiation dose received during treatment for either HL or testis cancer (LB Travis, personal communication). An exception to these trends is thyroid cancer risk, which has been shown to decrease after doses above 20 to 30 Gy in childhood cancer survivors.71,72 The radiation dose-response relation in SPC development, therefore, appears to be tissue specific. A recent systemic review on data on the radiation dose–response relationship for 11 second solid cancers found that for most SPCs (except for thyroid cancer), the dose-response curve was linear, even at doses of 60 Gy or higher.73
In addition to radiation dose, radiotherapy fields, which directly reflect the volume of normal exposed tissue, can affect SPC risk. The association between more extensive treatment fields and higher SPC risk has been shown in survivors of HL,74,75 breast cancer,76,77 and prostate cancer.78
Radiotherapy techniques and delivery systems have undergone major transformations over the past few decades.9 In recent years, highly conformal radiation techniques, including intensity-modulated radiation therapy (IMRT), have been adopted.79 IMRT uses sophisticated computer technology and dose optimization algorithms to produce precisely shaped dose distributions via multileaf collimator systems resulting in improved dose conformality in the high-dose region. However, with the use of multiple modulated beams, a larger volume of normal tissue is exposed to low doses of radiation. In addition, more radiation output, or monitor units (MU), are needed to deliver a specified dose to the target than for conventional treatments.80 Figure 143.2 illustrates the differences in dose distribution using conventional three-dimensional (3D) radiation therapy with anterior–posterior beam versus IMRT approach in a patient with mediastinal disease. The IMRT technique allows improved conformality at high doses, but at the expense of a larger volume of normal tissue exposed to low doses.
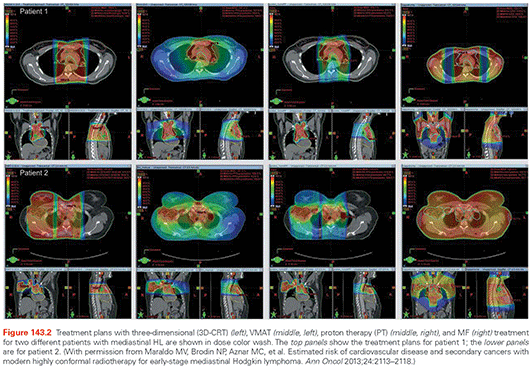
There is also increasing momentum for the use of proton therapy, given the physical characteristics of the depth dose curve,81,82 which peaks at the end of the particle range (Bragg peak). The beam has sharp edges with little side scatter, and the dose drops to zero after the Bragg peak. In addition, the integral dose, or the mean absorbed dose multiplied by the mass of irradiated tissue, is significantly lower with proton than photon beam. However, there have been concerns with regard to the SPC risk associated with ancillary neutron production, especially with the passive scattering technique, which results in dose exposure to the whole body.83,84 The newer beam scanning technique holds promise for reducing SPC risk as a result of low neutron contamination.83,85 This technique utilizes magnetically scanned pencil beams over the target volume, does not require scattering devices, collimation, or compensation, and therefore, is associated with reduced secondary neutron production from the treatment head.
There has been a growing literature on the estimation of SPC risks using radiobiologic modeling of SPC risk with these new radiotherapy techniques and delivery systems,80,86 some of which focused on specific disease sites, including prostate cancer,87–90 HL,91,92 gynecologic malignancies,93 lung cancer, and childhood malignancies.90,94 The estimated risks vary considerably depending on the model assumptions.88,89,92 Several studies estimated the lowest SPC risk with the scanning proton beam technique.91,94,95 Epidemiologic data for SPC risks in patients treated with modern techniques are currently lacking. Preliminary epidemiologic data on SPC risks following proton beam therapy for 503 patients treated at the Harvard Cyclotron with proton radiotherapy with passive scattering were reported.96 Compared to a matched cohort of 1,591 photon-treated patients identified in the SEER database,96 treatment with photon therapy was associated with a significantly increased risk of SPC (hazard ratio, 2.73; p <0.0001) after adjusting for age and gender. The median duration of follow-up was 6.1 and 7.7 years, respectively, in the photon and proton cohorts. Additional observation time may be needed to fully assess the long-term SPC risks following proton therapy. In a recent report issued by the National Council on Radiation Protection and Measurements (NCRP),9 recommendations with regard to future research on treatment-associated SPCs were outlined and are summarized in Table 143.1.

Chemotherapy
The association between alkylating agents and acute myeloid leukemia (AML) has long been recognized. Alkylating agents with known or suspected leukemogenic effects include mechlorethamine, chlorambucil, cyclophosphamide, melphalan, semustine, lomustine, carmustine, procarbazine, prednimustine, busulfan, dihydroxybusulfan, and the platinating agents.97–100 Of these, the platinating agents are the most commonly used group of cytotoxic drugs worldwide, and the most recently identified class of drugs for which highly significant dose-response relationships with therapy-associated leukemia have been observed.101,102 Chemotherapeutic topoisomerase II poisons, such as the epipodophyllotoxins (e.g., etoposide), the anthracyclines (e.g., epirubicin), and anthracenediones (e.g., mitoxantrone), are associated with a clinically and cytogenetically distinct type of AML, which show a relatively short induction period (median 2 to 3 years) and typically present without preceding myelodysplastic syndrome (MDS).103–105
Although the risk of treatment-related leukemias following chemotherapy has long been established, the induction of solid tumors is less well understood. Elevated risks of cancers of the bladder,106 bone,107 the lungs,53 gastrointestinal sites,66,108 and the thyroid109 following chemotherapy have been reported. There are also suggestions of associations between anthracyclines and excess sarcomas and thyroid cancers.108,109 Wilson et al.110 reported a 3.5-fold increased risk of renal cell carcinoma, albeit based on four cases, after cisplatin exposure in childhood cancer survivors. More recently, Fung et al.111 found a significant threefold (standardized incidence ratio [SIR] 3.37; 95% confidence interval [CI], 1.798 to 5.77; n = 13 cases) risk of renal cell carcinoma in a population-based study of 6,013 testicular cancer survivors treated in the modern cisplatin-based era (also see Testicular Cancer section). Just as the study of chemotherapy-induced leukemia has yielded considerable insight into the molecular mechanisms underlying SPC etiology, it will also be important to determine whether these or other mechanisms might be operant in second solid tumors.
Other agents used in cancer therapy may also have leukemogenic and carcinogenic effects. Immunosuppressants, including azathioprine, a thiopurine pro-drug related to 6-mercaptopurine and 6-thioguanine, is associated with significant excesses of AML,112 where risk is modified by host genetics (discussed in the Genetics section). In addition, azathioprine is also linked to the development of non-Hodgkin lymphoma (NHL),113 skin cancer,114 and brain tumors.115 There is evidence that granulocyte colony–stimulating factors, which are used in the treatment of lymphoblastic leukemia, breast, and other conditions, may also be associated with an increased risk of therapy-related AML.116–119 Similarly, the tyrosine-kinase inhibitors, such as imatinib, which is used to treat Philadelphia chromosome–positive cancers, including leukemia and gastrointestinal stromal tumors, might also be leukemogenic.120–123
Cancer after Bone Marrow Transplantation
Cancer after bone marrow transplantation is reviewed in Chapter 101. In brief, in survivors of high-dose therapy with bone marrow or stem cell transplantation, the estimated 10-year incidence of MDS/AML is 5% to 20%.124–126 Risk factors include the use of total body irradiation (TBI) in the conditioning regimen, prior radiotherapy, extent and type of prior chemotherapy, peripheral stem cell source, and low stem cell dose. Increased solid tumor risks are also evident after bone marrow or stem cell transplantation.127–129 Friedman et al.130 reported a 25-year cumulative incidence of breast cancer of 11% among 3,337 female 5-year survivors of allogeneic hematopoietic stem cell transplantation. In a multivariable analysis, risk factors included time since transplantation, use of TBI, and age <18 years at transplant. Rizzo et al.131 found a 3.3% 20-year cumulative risk of solid tumors in 28,874 recipients of allogeneic stem cell transplantation. The use of TBI as conditioning was significantly associated with solid tumor risk, but was restricted to patients age <30 years at transplant. The risk of invasive solid cancers among 4,318 allogeneic hematopoietic cell transplant (HCT) recipients given high-dose busulfan–cyclophosphamide without total body irradiation was 1.4-fold (95% CI, 1.08 to 1.79).132 Chronic graft-versus-host disease was an independent risk factor for all solid cancers, especially those of oral cavity.
GENETIC SUSCEPTIBILITY TO SECOND PRIMARY CANCERS
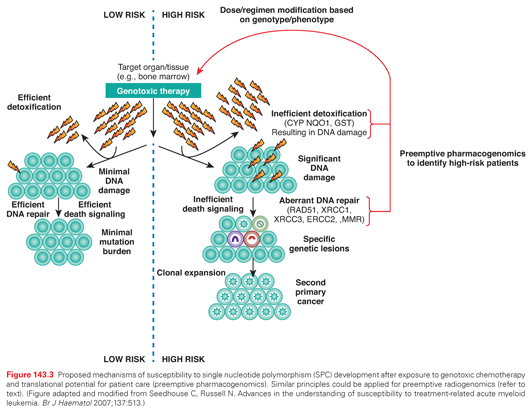
The role of chemotherapy and radiation in the development of SPC is well-established,9,10 although the existence of considerable interindividual variability suggests a role for genetic variation. Proposed mechanisms for susceptibility to the development of SPC after exposure to genotoxic therapy are summarized in Figure 143.3. SPC risk could potentially be modified by mutations in high-penetrance genes that lead to serious genetic diseases such as Li-Fraumeni syndrome,133 Fanconi anemia,134–137 and RB (see Pediatric Malignancy section). In fact, clues to the identification of loci important in affecting the risk of SPCs were initially suggested from studies of constitutional human cancer susceptibility syndromes (e.g., neurofibromatosis [NF]), many of which confer acute sensitivity to the mutagenic and cytotoxic effects of chemotherapy and radiotherapy, and where elevated risks of SPC are known or suspected.47,138 Consistent with this hypothesis, whole-genome sequencing of a patient with therapy-related AML identified a novel constitutional heterozygous deletion of TP53, affecting exons 7 through 9 encoding the DNA-binding domain of the protein. This likely represents a founder mutation in this patient with first primary breast and ovarian cancers and is consistent with a Li-Fraumeni syndrome–like phenotype.139 However, because of their low frequency, these alleles appear to contribute to only a small fraction of SPC risk at the population level. Nevertheless, this case highlights the possibility that TP53 and other established cancer susceptibility genes might harbor other novel low-frequency alleles conferring susceptibility to therapy-induced SPC, either with or without associated familial susceptibility to disease.
Evidence suggests that outside the context of familial cancer susceptibility syndromes, the genetic contribution to chemotherapy and radiotherapy-related cancer risk is multigenic, and likely related to the coinheritance of common low-penetrance polymorphisms in genes that regulate the availability of active drug metabolites, or those responsible for DNA damage signaling and repair. In the multigenic model, penetrance for SPC is predicted to be modulated by gene exposure and gene–gene interactions. For example, Ellis et al.140 utilized a case-control study design (171 cases) to examine the association of t-MDS/AML with two common functional p53-pathway variants—the mouse double minute 2 homolog (MDM2) SNP309 and the TP53 codon 72 polymorphism.140 Although neither polymorphism demonstrated a significant association, an interaction was detected such that patients with both MDM2 G and TP53 pro alleles were at increased risk of chemotherapy-related t-MDS/AML (interaction term p = 0.009). MDM2 is an E3 ubiquitin-protein ligase that recognizes the N-terminal transactivation domain of TP53 and negatively regulates its transcriptional activity in response to DNA damage. As such, interaction at the genetic level to modify SPC risk is plausible. Strengths of this study included the utilization of discovery (n = 80) and replication (n = 91) case sets. However, the investigators utilized healthy controls as a comparison group, thus precluding the ability to assess whether case-control differences reflected differences in susceptibility to primary disease or t-MDS/AML.
Genetic variation contributes 20% to 95% of the variability in cytotoxic drug disposition.141 Polymorphisms in genes involved in drug metabolism and transport are relevant in determining both disease-free survival and drug toxicity.142 Variation in DNA damage signaling and repair also plays a role in susceptibility to de novo cancer,143–145 and likely modifies SPC risk after exposure to DNA-damaging agents. Just as the pharmacology of cancer chemotherapy is impacted by underlying principles of pharmacokinetics, pharmacodynamics, and pharmacogenomics, these influences likely also contribute to SPC risk. Many loci that affect SPC risk encode proteins that metabolize and detoxify human carcinogens in an exposure-specific manner.146–149
Drug-Metabolizing Enzymes—Activation/Detoxification Pathways
The metabolism of genotoxic agents occurs in two phases. Phase I involves activation of substrates into highly reactive electrophilic intermediates that can damage DNA, a reaction principally performed by the cytochrome P-450 family of enzymes. Phase II enzymes (conjugation) function to inactivate genotoxic substrates, and include glutathione S-transferase (GST), NAD(P)H:quinone oxidoreductase-1, and others. The high activity of a phase I enzyme and the low activity of a phase II enzyme can result in DNA damage from excess harmful products (see Fig. 143.3).
Cytochrome P-450 Enzymes
The chemotherapeutic substrates of cytochrome P-450 proteins include cyclophosphamide, ifosfamide, thioTEPA, doxorubicin, and dacarbazine.150 The expression of these enzymes is highly variable among individuals because of several functionally relevant genetic polymorphisms, some of which are suspected to impact on SPC risk in patients treated with DNA damaging chemotherapy for a first primary cancer.146,147
Glutathione S-Transferases
Polymorphisms exist in cytosolic subfamilies such as μ [M], π [P], and θ [T]. GSTs detoxify doxorubicin, lomustine, busulfan, chlorambucil, cisplatin, cyclophosphamide, melphalan, and others.151 A common functional polymorphism in GSTP1 is associated with a significantly increased risk of developing therapy-related AML after exposure to chemotherapy that includes a leukemogenic GSTP1 substrate (chlorambucil, cyclophosphamide, melphalan, or doxorubicin) (odds ratio [OR], 4.34; 95% CI, 1.43 to 13.20), but does not influence the risk of radiogenic leukemia.152 Fabiani and colleagues153 also report an association with therapy-related myeloid neoplasia (n = 94 case) in carriers of the GSTP1 codon 105 risk allele (OR, 1.60; 95% CI, 0.95 to 2.69; p = 0.08), although findings were not stratified by treatment modality or exposure to a GSTP1 substrate.
DNA Repair
Variants in pathways that mediate cellular response to genotoxic damage are also likely to influence the risk of chemotherapy and radiotherapy-induced cancer, such as cell death signaling, cell proliferation, and DNA repair (MLH1,154 MSH2,155 XPD,156 XRCC1,157 XRCC3,158 NBN,158 RAD51,159–161 and P73162). DNA repair mechanisms protect cells from mutations in tumor suppressor genes and oncogenes that can lead to cancer initiation and progression, with an individual’s DNA repair capacity largely genetically determined.163 A number of DNA repair genes contain polymorphic variants, resulting in large interindividual variation in repair capacity, which could affect SPC risk.163 In support of this, a study of HL survivors suggested that those who developed a SPC had higher baseline levels of DNA damage than those who remained cancer free.164
Mismatch Repair
Mismatch repair (MMR) functions to correct mismatched DNA base pairs that arise as a result of misincorporation errors that avoided polymerase proofreading during DNA replication.165 Defects in this pathway result in genetic instability or a mutator phenotype, manifested by an elevated rate of microsatellite instability. Up to 90% of therapy-related AML patients have microsatellite instability, associated with methylation of the MMR family member MLH1,166,167 low expression of MSH2,168 or polymorphisms in MSH2.155,169
Double-Strand Break Repair
High levels of double-strand breaks follow ionizing radiation and chemotherapy exposures. Cellular pathways available to repair double-strand breaks include homologous recombination, nonhomologous end joining, and single-strand annealing.170 RAD51 is one of the central proteins in the homologous recombination pathway,171 and a common variant in the RAD51 gene (G-135C) is significantly overrepresented in patients with therapy-related AML compared with controls (C allele: OR, 2.7; 95% CI, 1.17 to 6.02).159 XRCC3 also functions in the homologous recombination double-strand break repair pathway by directly interacting with and stabilizing RAD51,172 and functions to maintain genetic stability.173,174 A polymorphism at codon 241 in the XRCC3 gene results in a Thr→Met amino acid substitution.175 The variant Met-encoding allele has been associated with a higher level of DNA damage, and has also been associated with increased levels of chromosome deletions in lymphocytes after radiation exposure,176 implying aberrant repair.177 Although the XRCC3 Thr241Met variant was not an independent risk factor for t-MDS/AML, a synergistic interaction with the RAD51-135C variant resulted in an eightfold increased risk of therapy-related AML (OR, 8.1; 95% CI, 2.2 to 29.7).159 Genes encoding proteins responsible for signaling DNA strand breaks are also predicted to carry risk alleles for therapy-induced SPCs. Prime candidates include those encoding components of the MRE11/RAD50/NBS1 (MRN) heterotrimeric complex responsible for recognition of DNA double-strand breaks in mammalian cells. Consistent with this hypothesis, the Women’s Environmental, Cancer, and Radiation Epidemiology (WECARE) study (a population-based, multicenter, case-control study of 708 women with asynchronous bilateral breast cancer and 1,395 women with unilateral breast cancer) identified a haplotype encompassing the RAD50 gene associated with the risk of contralateral breast cancer.178 The report identified an interaction with radiation dose (interaction term p = 0.006), where carriers of the RAD50 risk haplotype exposed to ≥1 Gy had an increased risk of contralateral breast cancer compared to unexposed carriers (rate ratio 4.31; 95% CI, 1.93 to 9.62). Taken together, these studies highlight the importance of gene–gene and gene exposure interactions in studies of SPC.
Base Excision Repair
Base excision repair corrects individually damaged bases that result from ionizing radiation and exogenous xenobiotic exposure. A common variant at codon 399 of the XRCC1 protein, which is central to the base excision repair pathway and functions as a scaffold for other repair components,179,180 was associated with an increased risk of developing t-AML,157 and has also been implicated as a risk factor in numerous other cancers.
Nucleotide Excision Repair
Nucleotide excision repair removes structurally unrelated bulky DNA lesions induced by radiation and chemotherapy. The nucleotide excision repair pathway is linked to transcription, and components of the pathway comprise the basal transcription factor IIH complex, which is required for transcription initiation by RNA polymerase II. Polymorphic variation in ERCC2, a component of the transcription factor IIH complex, has been associated with an increased risk of therapy-related AML.156
Hypothesis-Generating Genome-wide Association Studies
The associations reported previously were identified based on known gene-exposure interactions and analysis of loci in relevant pathways. Unfortunately, stratification by exposure is not always possible, particularly when a biological role has not been defined or when gene-exposure interactions are speculative. In this case, the application of high-density single nucleotide polymorphism arrays or next-generation sequencing can prove useful in identifying genetic variants and cellular pathways potentially important in causing chemotherapy- and radiotherapy-related cancer,139,140,181,182 as have genome-wide and whole transcriptome-based studies in laboratory animals.183,184
Knight et al.181 utilized a case-control study design to conduct a GWAS in patients who developed therapy-related leukemia (cases) and healthy controls. The discovery set included 80 cases and 150 controls. The relevant findings were replicated in an independent set of 70 cases and 95 controls. The investigators identified three SNPs (rs1394384 [OR, 0.29; 95% CI, 0.15 to 0.56], rs1381392 [OR, 2.08; 95% CI, 1.29 to 3.35], and rs1199098 [OR, 0.46; 95% CI, 0.27 to 0.79]) to be associated with t-MDS/AML with chromosome 5/7 abnormalities. The SNP rs1394384 is intronic to ACCN1, a gene encoding an amiloride-sensitive cation channel that is a member of the degenerin/epithelial sodium channel; rs1199098 is in linkage disequilibrium with IPMK, which encodes a multikinase that positively regulates the prosurvival protein kinase B (AKT) kinase and may modulate WNT/beta-catenin signaling; rs1381392 is not located near any annotated genes, miRNAs, or regulatory elements, although it lies in a region recurrently deleted in lung cancer. Although the investigators were able to confirm findings in an independent replication cohort, the utilization of a noncancer healthy control group raises concerns about any case-control differences being generated by the genetics of the primary cancer, rather than t-MDS/AML. Nevertheless, this study highlights the potential of GWAS to reveal novel risk alleles for SPC. Moreover, the identification of risk alleles unique to t-MDS/t-AML with somatic chromosome 5/7 abnormalities suggests that some risk alleles for SPCs may be subtype specific. Indeed, subtype-specific risk alleles have been identified for some first primary cancers. For example, Papaemmanuil and colleagues185 identified a risk allele at the ARID5B gene for pediatric B-cell precursor acute lymphoblastic leukemia that appears to be selective for disease with somatic hyperdiploidy. Likewise, there is compelling evidence that estrogen receptor-positive and estrogen receptor-negative breast cancers have different genetic components to their etiology.186,187 These data highlight the importance of appropriate stratification to identify the context (e.g., gender, exposure, disease subtype) in which risk alleles for SPCs are operating and where penetrance for SPC is highest, an approach that will facilitate translation into the clinic for patient benefit.
Considering this model, Best et al.52 performed a GWAS to identify genetic variants associated with radiation-associated solid malignancies in HL survivors. They identified two variants at chromosome 6q21 associated with SPC, but which only modified risk in patients who were exposed to radiation in childhood and had no penetrance for SPC when exposure occurred in adulthood. The variants comprise a risk locus associated with decreased basal expression of PRDM1 and impaired protein induction after radiation exposure. These data suggest a novel gene-exposure interaction implicating PRDM1 in the etiology of radiation therapy-induced SPC, with evidence that age at radiation therapy exposure modifies the association between variants in this gene and SPC risk. Hosking and colleagues51 also conducted a GWAS for inherited susceptibility to radiation-associated malignancy—specifically, meningioma in patients previously treated for tinea capitis. Although the average radiation dose to the brain was relatively low (~1.5 Gy), there was a significant increase in the risk of meningioma in exposed individuals (excess relative risk [ERR]/Gy 4.63; 95% CI, 2.4 to 9.1).188 The high level of natural linkage disequilibrium in the exposed population allowed the authors to identify putative risk haplotypes associated with radiation-induced meningioma at 18q21.1 (p = 7.5 × 10−5), 18q21.32 (p = 2.8 × 10−5), and 10q21.3 (p = 1.6 × 10−4), although none reached GWAS significance and they require validation in independent cohorts with the inclusion of appropriate control groups to exclude an association with meningioma without a radiation etiology. To the extent that carcinogenic pathways operant at very low doses of radiotherapy may also be functional at the high doses given in cancer treatment, these genomic regions may also harbor risk alleles predisposing to meningioma in cancer survivors who were administered cranial radiotherapy.
Although most candidate gene association studies for SPC have focused on pathways involved in regulating cell death or maintaining genetic stability in response to genotoxic therapies, genome-wide approaches also have the potential to uncover risk alleles in novel pathways influencing the risk of therapy-induced malignancy. By conducting a genome-wide linkage study using inbred mice, Boulton et al.189 identified genomic regions harboring putative risk loci for radiogenic AML. One such locus on chromosome 1 encompassed Scfr1, a gene encoding a key regulator of stem cell frequency, suggesting that the size of the stem cell pool at risk of transformation is genetically defined and a determinant of radiation-induced AML. If validated in human disease, these data would add another dimension to risk models of therapy-induced SPC.
Preemptive Pharmacogenomics and Preemptive Radiogenomics
Regardless of whether identified via candidate gene, genome-wide, or systems biology approaches, accurate estimates of the contribution of single gene variants will require large studies with sufficient power such that specific gene–gene or gene exposure can be identified. Indeed, the identification of the context in which penetrance for a genetic variant for SPC is greatest will facilitate the identification of patients at largest risk. Moreover, precise estimates of the magnitude of the effect(s) are required in order to develop personalized risk-adapted clinical strategies, including dose modification, alternative treatments, and posttherapy surveillance. Risk management could prove particularly important in children or young adults with primary cancers in whom cure rates are high and the risk of a subsequent cancer is associated with substantial morbidity or premature mortality.
In several instances, such as the relation between constitutional thiopurine methyltransferase (TPMT) activity and SPC risk, the goal of clinical translation has already been achieved. For example, children with acute lymphoblastic leukemia (ALL) who received combined-modality therapy that included 6-mercaptopurine at a dose of 75 mg/m2 and who had high TPMT activity (conferred by constitutional polymorphism in the TMPT gene) had an 8.3% cumulative risk of subsequently developing a brain tumor, whereas children with low TPMT activity had a 42% risk of a posttherapy brain tumor (p = 0.0077).115 It was hypothesized that the deficient TPMT activity resulted in higher exposures to thioguanine nucleotide metabolites of 6-mercaptopurine during the period of radiation. Likewise, patients with lower TPMT activity also had an increased risk of developing AML and other SPC after high-dose thioguanine chemotherapy.190–192 However, a reduction in 6-mercaptopurine dose (50 mg/m2), along with other changes adopted as part of the Berlin-Frankfurt-Munster protocol for the treatment of childhood ALL, appeared to attenuate the effect of TPMT status on risk of AML after thioguanine-containing chemotherapy.193
The gene-exposure interaction between TPMT and chemotherapeutic thioguanine nucleotides is well-established and strongly associated with several life-threatening toxicities (bone marrow, hepatotoxicity) in cancer patients inadvertently overtreated with these agents as a consequence of genotype. Indeed, convincing evidence of acute toxicity in TPMT-compromised patients prompted the U.S. Food and Drug Administration (FDA) to add a warning to the drug along with information on genotype frequencies and a recommendation for TPMT genetic testing following the administration of 6-mercaptopurine for patients with clinical or laboratory evidence of severe bone marrow toxicity, particularly myelosuppression.194 Evidence of elevated SPC risk in carriers of TPMT null alleles115,190–192 also underscores the importance of preemptive genotyping within this context, rather than posttherapy response-mode genotyping, particularly because there is now convincing evidence that toxicity and SPC risk can both be ameliorated with appropriate dose modification.193 Preemptive testing for TPMT genotype and/or phenotype status is recommended by the Clinical Pharmacogenetics Implementation Consortium and others prior to initiating therapy that includes thioguanine nucleotides, including 6-mercaptopurine.192,195–201
Although it remains to be determined whether the TPMT model can be extended to other gene exposure interactions involving therapeutic agents, dose, and/or regimen modification has the potential to reduce the risk of SPCs in genetically susceptible individuals. Indeed, this principle has also been applied to other populations at risk of therapy-induced SPC, such as patients with NF1. Neurofibromatosis (NF) (caused by mutations in the NF1 gene) is an autosomal dominantly inherited disorder characterized by a predisposition to benign tumors of the nerve sheath. There is evidence from studies of both laboratory animals202 and humans203 that constitutional heterozygosity for NF1 pathogenic mutations confers susceptibility to radiogenic cancer, such that many investigators now recommend avoidance of radiotherapy whenever possible to treat tumors in NF patients.204,205
Preemptive testing of either genotype or phenotype status, such that appropriate dose modification or alternative treatment approaches can be implemented at first treatment, is critical to reducing therapy-induced SPC risk in patients with a strong genetic predisposition. Although not routinely yet considered in the context of SPC risk, preemptive testing based on gene-exposure interactions could potentially be applied in other clinical settings, and the value of this approach as a mechanism to reduce acute adverse drug events is already evident.206,207 TPMT and NF1 represent two examples where dose/regimen modification can ameliorate the risk of therapy-induced SPC in affected individuals, providing proof of principle of the potential value of preemptive genomics to reduce the risk of therapy-induced malignancy in selected cancer survivors (see Fig. 143.3). Moreover, as additional data are accrued with regard to molecular mechanisms that underlie radiotherapy-associated carcinogenesis,9,10,52,208 the principles previously outlined can also be expanded to introduce a new field of preemptive radiogenomics.
RISK OF SECOND MALIGNANCY IN PATIENTS WITH SELECTED PRIMARY CANCERS
Lymphoma
The use of combined-modality therapy in patients with both HL and NHL has resulted in significant improvements in survival, but with the attendant risk of SPCs, including leukemia and a number of solid tumors.209
Hodgkin Lymphoma
Reports from large, well-characterized cohorts of HL survivors show that the risk of SPC is 2 to 18 times higher than in the general population.55,56,210–218 Dores et al.216 reported an analysis of 32,591 HL survivors (1935 to 1994) in 16 population-based registries. A total of 2,153 SPCs (relative risk [RR] = 2.3) was observed, including 1,726 solid tumors (RR = 2.0). The 25-year cumulative incidence of a solid tumor was 21.9%. Temporal trends for cancers of the female breast, thyroid, esophagus, stomach, rectum, bladder, and bone/connective tissue were suggestive of a radiogenic effect.
Hodgson et al.54 reported on 18,862 5-year survivors of HL in 13 population-based registries from North America and Europe, treated between 1970 and 2001, and identified 1,490 cases of solid tumors (RR = 2.38). Cancers of the lung, breast, and gastrointestinal sites comprised the three most common diagnoses accounting for 21%, 19%, and 17% of the cases, respectively. The RRs of lung and breast cancers were 6.7 and 6.1, respectively, and the RR of gastrointestinal cancers ranged from 1.8 for anorectal cancer to 9.5 for stomach cancer. Other solid tumors with significantly increased risks included cancers of the head and neck, kidney, thyroid, gynecologic organs, soft tissue, bone, and brain.
It is important to recognize that radiation therapy for HL has undergone substantial changes in 4 decades with fields decreasing from total nodal irradiation to involved-field radiation therapy and, most recently, to involved site therapy. Prescribed dose has also decreased from 40 to 44 Gy to as low as 20 Gy in selected early-stage patients as part of combined modality therapy. Modeling studies show significant reduction in SPC with lower dose and smaller fields and with modern radiation therapy techniques. Many currently available epidemiologic studies of SPC after HL were based on patients treated in the earlier era, and long-term follow-up of patients treated with modern techniques will be needed to accurately assess SPC risk.
Non-Hodgkin Lymphoma
Several population-based studies have provided estimates of the risk of SPCs following NHL.91–95,219–223 In a study by Dores et al.,223 5,490 subsequent cancers were observed among 73,958 2-month survivors of NHL, reflecting an overall elevated risk of 14% compared with the general population (observed-to-expected ratio [O/E] = 1.14; 95% CI, 1.11 to 1.7; excess absolute risk [EAR] = 19). The cumulative risk of SPC reached 12.3% within 25 years. Lung cancer accounted for the largest excess of SPCs after NHL. Results from the SEER program223 corroborated findings in previous surveys of NHL, which reported significant excesses of cutaneous melanoma, Kaposi’s sarcoma, HL, AML, and cancers of the buccal cavity, lip, lungs, urinary bladder, and kidneys.219–221,224
Radiotherapy for NHL appeared related to the increased risks for bladder cancer, bone sarcoma, and mesothelioma.223 Chemotherapy for NHL has been associated with an increased risk for leukemia225 as well as several solid tumors, including bladder cancer106 with dose-dependent relationships. The immunosuppressive state associated with NHL226 may contribute to excesses reported for cutaneous melanoma, HL, and lip cancer.223,224,227,228 Most recently, analyses of the SEER program data reported SPC risk by major histologic groups of NHL,229 akin to studies of SPC after ovarian cancer.230 Among patients without known HIV/AIDS, the SIR of SPC after chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL) were 1.19 (95% CI, 1.13 to 1.25), 1.11 (95% CI, 1.04 to 1.19), and 1.03 (95% CI, 0.96 to 1.11), respectively. Risks of lung cancer and melanoma were significantly increased in survivors of CLL/SLL and FL, but not DLBCL. Moreover, an elevated risk was observed regardless of whether radiation therapy was given as part of the initial treatment. An increased risk of acute nonlymphocytic leukemia was seen after FL and DLBCL, with a SIR of 4.96 (95% CI, 3.87 to 6.87) and 5.96 (95% CI, 4.15 to 8.29), respectively, but not after CLL/SLL. The increased leukemia risk was restricted to survivors who received chemotherapy initially.
In summary, NHL survivors are at increased risk for a number of SPCs, although to a considerably lesser degree than HL patients. The persistent increase in the risk of all SPCs for more than 20 years after NHL treatment alerts clinicians to the importance of long-term surveillance.
Selected Second Primary Cancers Following Hodgkin Lymphoma or Non-Hodgkin Lymphoma
Breast Cancer
Breast cancer is one of the most common solid tumors after HL. Studies have consistently shown that young age at radiotherapy is associated with a significantly higher risk of breast cancer.54–57,231 In a population-based cohort study,54 the absolute risks of breast cancer in women diagnosed with HL at ages 15 through 25 years were higher than the absolute risks of women in the general population between 50 and 54 years, a standard age when mammography screening is recommended. Travis et al.232 developed estimates of the cumulative absolute risk of breast cancer, taking into account age and calendar year of HL diagnosis, current age, radiation dose, and competing causes of mortality. Thus, for a HL survivor who received a chest radiation dose of ≥40 Gy at age 25 years, the cumulative absolute risks of breast cancer at 10, 20, and 30 years were estimated to be 1.4%, 11% and 29%, respectively. A report from the Childhood Cancer Survivor Study (CCSS) estimated the risk of breast cancer in a cohort of 1,268 female 5-year childhood cancer survivors given chest radiation therapy. The overall cumulative incidence of breast cancer by age 50 years among HL survivors was 30% (95% CI, 25% to 35%).
There is a clear radiation dose-response relation for breast cancer risk after HL. In an international case-control study of breast cancer after HL that included 105 cases of breast cancer and 266 matched controls,61 radiation dose to the area of the breast where the tumor developed in the case (and a comparable area in controls) was estimated. Breast cancer risk increased significantly with increasing radiation dose to reach eightfold for the highest category (median dose 42 Gy) compared to the lowest dose group (<4 Gy) (p-trend for dose <0.001) (Table 143.2).
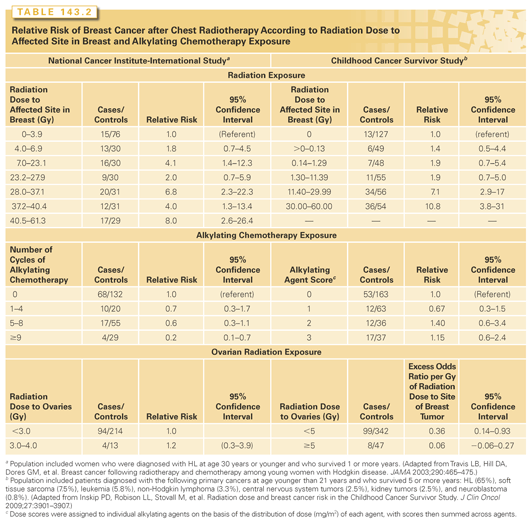
A significant radiation dose-response relation was similarly demonstrated in a Dutch study that was included in the previous international investigation.62 The CCSS also published a case-control study of 120 cases of breast cancer (65% in survivors of HL) matched to 464 controls.63 Again, a significant linear radiation dose-response was observed (p-trend <0.0001), with an estimated relative risk of breast cancer of 6.4 at 20 Gy and 11.8 at 40 Gy (see Table 143.2). Radiation field size for HL also affects breast cancer risk. A cohort study of 1,122 5-year female HL survivors showed that mantle field irradiation (inclusion of mediastinum, bilateral axillae and neck) was associated with a 2.7-fold increased risk (95% CI:, 1.1 to 6.9) of breast cancer compared with mediastinal irradiation alone.74 A meta-analysis of SPC after HL found that extended-field radiotherapy was associated with a significantly higher breast cancer risk than involved-field radiotherapy (OR = 3.25; p = 0.04).75 A study from Yale University evaluated solid tumor risks in 404 HL survivors and observed 10 cases of breast cancer in women who received radiation treatments that included the axillae, but no cases when the axillae were omitted from the field.
Early menopause has been shown to be protective against breast cancer in HL survivors treated with radiotherapy. In particular, the Dutch study clearly documented that breast cancer risk reduction associated with chemotherapy was due to premature menopause.62 In a more recent HL cohort study from the same group,74 among women age <41 years at treatment, those women with >20 years of ovarian function post-treatment had a significantly higher risk of breast cancer compared to patients with 10 to 20 years of intact ovarian function after therapy (hazard ratio = 5.3; 95% CI, 2.9 to 9.9).
Several studies have shown a reduced risk of breast cancer among NHL survivors compared to the general population, likely due to chemotherapy-related early menopause.233,234 Although breast cancer risk after mediastinal irradiation for NHL has not been specifically assessed, indirect data suggested an increased risk with younger age at irradiation. In a SEER-based study of 77,876 NHL patients,222 the relative risk of breast cancer was significantly increased among women irradiated at age <25 years (RR = 5.05; p <0.05), whereas breast cancer excesses were not observed in the entire irradiated cohort. Similarly, in a Swedish Cancer Registry–based study of 29,134 patients with NHL,235 the relative risk of breast cancer was significantly increased only among women diagnosed between age 20 to 39 years (RR = 3.29; 95% CI, 1.91 to 5.67) and not in older patients. The increased breast cancer risk in these young women may have been due to chest radiotherapy, although this cannot be confirmed because the sites of irradiation were not described.
Lung Cancer
Significantly elevated three- to sevenfold relative risks of lung cancer have been reported after HL,53–56,65,216,217,236 with a dose-dependent relationship documented for antecedent alkylating agent chemotherapy53,236 and for radiation dose to the area of the lungs in which the cancer developed.53 The modifying effect of smoking history on treatment-related lung cancer in HL survivors was investigated in a case-control study by Travis et al.53 Using patients who had minimal radiation or alkylating chemotherapy exposure and who were nonsmokers as the reference group, exposure to >5 Gy of radiotherapy and/or alkylating agent chemotherapy was associated with a 4.3- to 7.2-fold increased risk of lung cancer. The relative risk increased to 16.8 and 20.2, respectively, in patients who had either one of the treatment exposures and who used tobacco. Moreover, the relative risk was 49.1 in patients who had >5 Gy of radiotherapy, received alkylating chemotherapy, and who used tobacco, consistent with a multiplicative effect of tobacco use on the risk of treatment-related lung cancer.
The risk of lung cancer is modestly increased after NHL, with most studies reporting a risk of less than twofold compared to the general population.219,220,222,233–235,237,238 The increased risk appears to be associated with both chemotherapy and radiotherapy, with significantly increasing risk following higher cumulative doses of chemotherapy.237 Increased risks for malignant mesothelioma have been reported in survivors of both HL and NHL, although the excesses appear limited to irradiated patients.236,239,240
Gastrointestinal Malignancies
Population-based and multi-institutional cohort studies have consistently reported an increased risk, compared to the general population, for cancers of the esophagus, the stomach, the colon, and the rectum among HL survivors.55,56,215,216,241,242 Van den Belt-Dusebout et al.66 conducted a nested case-control study of 5,142 Dutch testicular cancer and HL survivors to examine the role of radiation dose and chemotherapy in the etiology of gastric cancer. Because age characteristics of the testicular cancer and HL patients were generally similar, these populations were pooled, although it should be kept in mind that the underlying cancers are distinctly different and that only males were represented in the former group. A 3.4-fold increased risk of gastric cancer was observed compared with the general population. Risk increased with increasing mean radiation dose to the stomach, with an ERR of 0.84 per Gy. Adjusted for radiation, higher doses of procarbazine (≥13,000 mg versus <10,000 mg) were associated with a 5.4-fold increased risk. Smoking more than 10 cigarettes per day was associated with a 1.6-fold increased risk compared with survivors who did not smoke.
In a nested case-control study of stomach cancer risk in 19,882 HL survivors,243 89 cases and 190 matched controls were included, with 19 of the cases and 69 controls previously reported.66 Risk was significantly associated with radiation dose to the stomach (excess OR, 0.09/Gy; p-trend <0.001) and number of cycles of alkylating chemotherapy (p-trend = 0.02) Patients who received gastric radiation doses of 25 Gy or more to the stomach and cumulative doses of procarbazine of 5,600 mg/m2 or higher had an OR of 77.5 (95% CI, 14.7 to 1,452). Exposure to dacarbazine was also associated with an increased stomach cancer risk (OR, 8.8; 95% CI, 2.1 to 46.6), after adjustment for radiation and procarbazine dose.
Breast Cancer
Women with breast cancer are at significantly elevated risk for developing SPCs. The largest amount of data have accrued on the risk of contralateral breast cancer (CBC), which is estimated to be increased two- to fivefold. Factors that may modify the increased risk include preexisting breast cancer risk factors such as genetic predisposition, reproductive factors and health habits, as well as treatment exposures.244–247
Conflicting data exist on the contribution of radiotherapy to excess risks of CBC.248–251 In a case-control study by Boice et al.,250 the overall relative risk of CBC was not significantly increased after radiotherapy (RR, 1.19; 95% CI, 0.94 to 1.15). Among women age <45 years at the time of irradiation, however, the relative risk was significantly elevated at 1.59 (95% CI, 1.07 to 2.36). Hooning et al.58 assessed the long-term risk of CBC in 7,221 breast cancer survivors, and found a 2.91-fold (95% CI, 2.66 to 3.18) increased risk compared with the general female population (absolute excess risk [AER] = 46.1 out of 10,000 person-years). The risk of radiotherapy-related CBC also increased significantly with decreasing age at treatment; among women who received radiotherapy at age <35 years, the hazard ratio for CBC was 1.78 (95% CI, 0.85 to 3.72), whereas for women irradiated at age ≥45 years, risk decreased to 1.09 (95% CI, 0.82 to 1.45). The Early Breast Cancer Trialists’ Collaborative Group reported a significantly increased risk of CBC with radiotherapy, mainly during the period 5 to 14 years after randomization (RR, 1.43; p = 0.00001), and the increased risk was significant even among women aged ≥50 years when randomized (RR, 1.25; p = 0.002).252 In a report from the WECARE group,59 where absorbed doses to quadrants of the contralateral breast were estimated, women <40 years of age who received >1.0 Gy of absorbed dose to the specific quadrant of the contralateral breast had a significantly increased 2.5-fold greater risk for CBC than unexposed women. No excess risk was observed in women >40 years of age. In contrast, in an earlier case-control study from Denmark, a significant difference in CBC risk was not observed in women who did and did not receive radiotherapy, regardless of age at treatment.253 A study from the Institut Curie, which included 13,472 women also similarly showed no increased risk of CBC when comparing women who did or did not receive radiotherapy (RR, 1.1; 95% CI, 0.96 to 1.27). Currently available data, therefore, support a modest risk of CBC after radiotherapy, mostly limited to younger women at the time of treatment.
Several other solid tumors have been linked to radiotherapy for breast cancer, including lung cancer, sarcoma, and esophageal cancers. Compared to women who did not receive radiotherapy, women given radiotherapy have a 1.5- to 3-fold increased risk of developing lung cancer.248,254,255 In a case control study by Kaufman et al.,256 113 breast cancer patients who developed lung cancer were matched to 364 controls without lung cancer; excess risks of lung cancer following postmastectomy radiotherapy were limited to ever-smokers. Compared with nonsmoking women who did not receive postmastectomy radiotherapy, the adjusted OR for lung cancer among nonirradiated ever-smokers was 5.9 (95% CI, 27 to 12.8) and the adjusted odds ratio among ever-smokers who also received postmastectomy radiotherapy was 18.9 (95% CI, 7.9 to 45.4). Others have shown that the increased risks of lung cancer appeared more related to postmastectomy radiotherapy, in which the radiation target volume often also includes the supraclavicular, axillary, and/or internal mammary nodal regions, thus exposing a larger volume of lung tissue, whereas the risk after postlumpectomy radiotherapy is less clear.76,77 Similarly, in a population-based study by Zablotska et al.77 of squamous cell esophageal cancer after adjuvant radiotherapy for breast cancer, the relative risk was significantly increased only among women who received postmastectomy radiotherapy, but not among those who received postlumpectomy radiotherapy. In a recent international population-based nested case-control study of esophageal cancer in breast cancer survivors that included 252 cases and 488 controls, the esophageal cancer risk was 8.3-fold (95% CI, 2.7 to 28) higher in women who received doses of 35 Gy or higher, compared with women who received no radiation dose to the site of esophageal tumor.64
Although the occurrence of sarcoma after breast cancer is a rare event, with a 15-year incidence rate of <0.5%, the relative risk has been estimated to be as high as sevenfold, given the low background incidence in the general population.248,257,258 In a study by Rubino et al.,70 all sarcomas occurred among women initially given radiotherapy, and in all cases, the sarcomas were located in the irradiated fields or in the upper extremity of the arm ipsilateral to the treated breast. Further, a significant dose-response relationship was demonstrated. In a SEER-based study of 563,155 breast cancer patients, women who received radiotherapy had a 1.5-fold risk (95% CI, 1.3 to 1.8) of all types of sarcomas, and a 7.6-fold risk (95% CI,4.9 to 11.9) of angiosarcoma, compared to women who did not receive radiotherapy.259 In addition to radiotherapy, partial mastectomies and lymph node dissections were also independent risk factors for the development of angiosarcoma. Unlike other radiation-related soft tissue sarcomas, angiosarcoma has a short latency and can occur in the first 5 years after therapy.
Endometrial cancer after breast cancer can be linked with both shared etiologic factors and antecedent therapy—in particular, tamoxifen therapy. Several large studies have demonstrated a two- to fourfold increased risk of endometrial cancer after tamoxifen therapy.30 Earlier studies indicated that endometrial cancer after tamoxifen use may have a more favorable prognosis, although recent data have raised the concern that tamoxifen-related endometrial cancers may have a more aggressive behavior.260–262 In a study that included 732 women with endometrial carcinoma, 59 patients (8%) had a previous diagnosis of breast cancer, of whom 29 (49%) had used tamoxifen.263 A longer duration of tamoxifen use (60 versus 46 months; p = 0.034) was significantly associated with high-risk histologic type. A Dutch study, which examined endometrial cancer after tamoxifen treatment for breast cancer, found that long-term tamoxifen use was associated with a higher proportion of nonendometrioid tumors than nonusers (32.7% versus 17.4%; p = 0.004), especially serous adenocarcinomas and carcinosarcomas; a higher proportion of International Federation of Gynecology and Obstetrics (FIGO) stage III and IV tumors was also observed (20.0% versus 11.3%; p = 0.049).261 The differences in histologic and stage distributions translated to a significantly lower 3-year endometrial cancer-specific survival among long-term tamoxifen users (82% versus 93%; p = 0.001).
Testicular Cancer
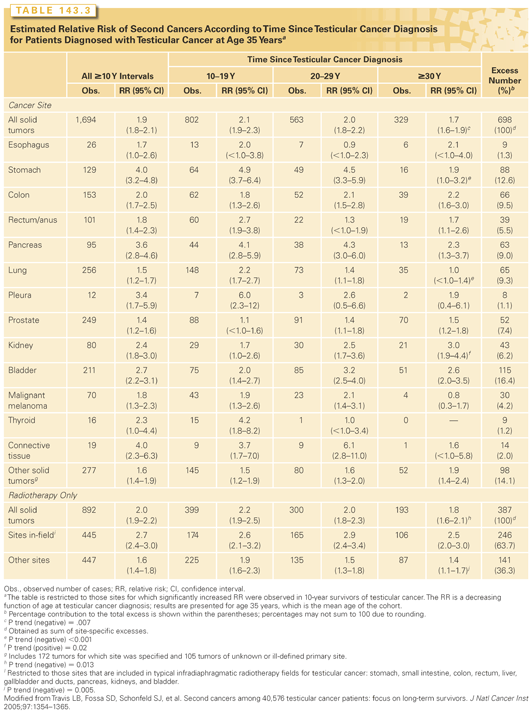
Significant excesses of solid tumors, leukemia, and contralateral testicular cancer (CTC) have been documented in testicular cancer survivors (TCS).102,264–275 In an international population-based survey of 40,576 TCS,264 significantly elevated 1.5- to 4-fold relative risks were noted for malignant melanoma and cancers of the lungs, thyroid, esophagus, pleura, stomach, pancreas, colon, rectum, kidneys, bladder, and connective tissue among 10-year survivors (Table 143.3). By age 75 years, men who were diagnosed with seminomas or nonseminomatous tumors at age 35 years experienced cumulative risks of solid cancer of 36% and 31%, respectively. Among TCS treated with radiotherapy alone, relative risks of SPC at sites included in typical infradiaphragmatic radiotherapy fields were significantly larger than risks at nonexposed sites (RR, 2.7 versus RR, 1.6; p <0.05) and remained elevated for ≥35 years. No decrease in the risk of postradiotherapy in-site SPC was observed for seminoma patients diagnosed between 1975 and 2001, although a lowered risk was noted among nonseminoma patients. In a case-control investigation of 23 stomach cancers among TCS,66 a significant relation with increasing radiation dose was observed. In the international series,264 the relative risk (RR = 1.8) of solid tumors was significantly elevated after chemotherapy alone, although information on specific cytotoxic drugs was not available. A subsequent investigation265 showed that cisplatin-based chemotherapy was associated with a significantly elevated twofold risk of solid tumors, confirming other reports.275
In a study exploring cause-specific mortality in patients with stage I seminoma using data from the SEER program, with 121,037 person-years of follow-up, the standardized mortality ratio (SMR) of SPC was 1.81 (95% CI, 1.61 to 2.03).276 The risk was higher among irradiated patients, with an estimated 15-year cumulative risk of 2.64%.
Cytotoxic drugs used to treat testicular cancer (TC) that have been related with excess secondary leukemias include etoposide and cisplatin.102,271,273,274 Kollmannsberger et al.274 estimated that the cumulative risk of leukemia for TCS given etoposide at total doses of <2,000 mg/m2 and >2,000 mg/m2 was 0.5% and 2%, respectively. A strong dose-response relation (p <0.001) between cumulative dose of cisplatin and subsequent leukemia risk was reported by Travis et al.,102 who also noted a nonsignificant threefold risk following involved-field radiotherapy. Fung et al.111 recently examined solid tumor risk after surgery and chemotherapy in 12,691 testicular nonseminoma survivors in the SEER program (1980 to 2008). A significantly increased RR of solid tumor of 1.43 (95% CI, 1.18 to 1.73) was found (median latency period: 12.5 years), with significantly increased site-specific risks for cancers of the kidney (SIR, 3.37; 95% CI, 1.79 to 5.77), thyroid (SIR, 4.40; 95% CI, 2.19 to 7.88), and soft tissue (SIR, 7.49; 95% CI, 3.59 to 13.78).
In a population-based study of 29,515 TCS, the 15-year cumulative risk of CTC was 1.9%, representing a 12.4-fold higher risk than expected in the general population.272 Chemotherapy may reduce the risk of CTC,277,278 although the delaying effect and duration of cisplatin-based chemotherapy on CTC incidence should be examined further.
In the future, it will be important to determine whether the decrease in radiation dose and field size in the 1990s,279,280 along with the use of carboplatin as adjuvant therapy in seminoma patients,281
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


