Sarcomas
Sarcomas are a complex and heterogeneous family of malignancies of mesenchymal origin. As opposed to carcinomas, which represent malignancies of epithelial tissues, sarcomas are neoplastic disorders that differentiate into lineages related to “connective tissues,” broadly including bone, fat, stromal supportive cells (such as fibroblasts), cartilage, and blood vessels. The term “sarcoma” is derived from the Greek root sarc (flesh) and the suffix – oma (tumor). Sarcasm, a flesh-tearing criticism, and sarcophagus, a carrier for a body, are based on the same root. FLOAT NOT FOUND FLOAT NOT FOUND
Though uncommon, sarcomas are highly informative about the mechanisms of human neoplasia. Virtually no class of human solid tumors has yielded as many pathogenetic clues as to specific molecular aberrancies linked to individual tumor subtypes as sarcomas. Tumor-specific chromosomal translocations have been defined for many different types of sarcomas. Identification of these molecular pathways of cancer has had both diagnostic and therapeutic impact. For example, the identity of Ewing sarcoma with primitive neuroectodermal tumor was identified because these entities shared a common cytogenetic abnormality. The best example of the therapeutic impact of this knowledge has been the development of imatinib mesylate (Gleevec; Novartis, Basel, Switzerland) and sunitinib malate (Sutent; Pfizer, New York, NY) as molecularly targeted therapy for gastrointestinal stromal tumors (GISTs), a subtype of sarcoma that was essentially resistant to any systemic therapies before the era of kinase-inhibiting agents such as these. It is hoped that with additional knowledge of oncogenic molecular mechanisms derived from sarcomas, accelerated translation of basic science will allow the development of rational, mechanism-based therapies for many other types of malignancies.
Sarcomas of soft tissue and bone currently represent 1% of adult malignancies but disproportionately affect children, composing 15% of pediatric malignancies. The sex predominantly affected differs considerably among the histologic variants. For example, 70% of patients with Kaposi sarcoma are male, as are 60% of those with liposarcoma and embryonal rhabdomyosarcoma, and 35% with leiomyosarcoma. Sarcomas develop in individuals of all ages. Embryonal rhabdomyosarcoma generally occurs in children and adolescents under 20 years of age; synovial sarcoma, osteosarcoma, alveolar rhabdomyosarcoma, and Ewing sarcoma arise in adolescents and young adults, whereas leiomyosarcoma, chondrosarcoma, and GISTs typically occur in individuals over 50 years of age.
Sarcomas are traditionally classified in two major groupings: those arising in bone or cartilage and those originating in soft tissue. Soft tissue sarcomas can be further subdivided based upon the anatomic site of origin: for example, those that develop in the extremities, retroperitoneum, or visceral organs (such as the gastrointestinal [GI] and gynecologic tracts) ( Tables 12.1 , 12.2 ).
| Primary Tumor (T) | |
|---|---|
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor 8 cm or less in greatest dimension |
| T2 | Tumor more than 8 cm in greatest dimension |
| T3 | Discontinuous tumors in the primary bone site |
| Regional Lymph Nodes (N) | |
|---|---|
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastasis (M) | |
|---|---|
| MX | Distant metastasis cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Lung |
| M1b | Other distant sites |
| Stage Grouping | |||||
|---|---|---|---|---|---|
| IA | T1 | N0 | M0 | G1,2 | Low grade |
| IB | T2 | N0 | M0 | G1,2 | Low grade |
| IIA | T1 | N0 | M0 | G3,4 | High grade |
| IIB | T2 | N0 | M0 | G3,4 | High grade |
| III | T3 | N0 | M0 | Any G | |
| IVA | Any T | N0 | M1a | Any G | |
| IVB | Any T | N1 | Any M | Any G | |
| Any T | Any N | M1 | Any G | ||
| Histologic Grade (G) * | |
|---|---|
| GX | Grade cannot be assessed |
| G1 | Well differentiated–low grade |
| G2 | Moderately differentiated–low grade |
| G3 | Poorly differentiated–high grade |
| G4 | Undifferentiated–high grade |
| Sarcoma Type | Frequency (%) |
|---|---|
| Bone | |
| Osteosarcoma | 45 |
| Chondrosarcoma | 22 |
| Ewing sarcoma | 13 |
| Chordoma | 9 |
| Fibrosarcoma | 7 |
| Malignant fibrous histiocytoma | 2 |
| Angiosarcoma | 1 |
| Other | 1 |
| Soft Tissue Sarcomas | |
| Liposarcoma | 25 |
| Well-differentiated (atypical lipomatous tumor) (45) | |
| Dedifferentiated liposarcoma (15) | |
| Myxoid (and round cell) liposarcoma (35) | |
| Pleomorphic liposarcoma (5) | |
| Leiomyosarcoma | 15 |
| Synovial sarcoma | 10 |
| Undifferentiated pleomorphic sarcoma (“MFH”) | 5–10 |
| Malignant peripheral nerve sheath tumor | 5–10 |
| Rhabdomyosarcoma | 5 |
| Myxofibrosarcoma | 5 |
| Other | 20 |
SARCOMAS OF BONE AND CARTILAGE
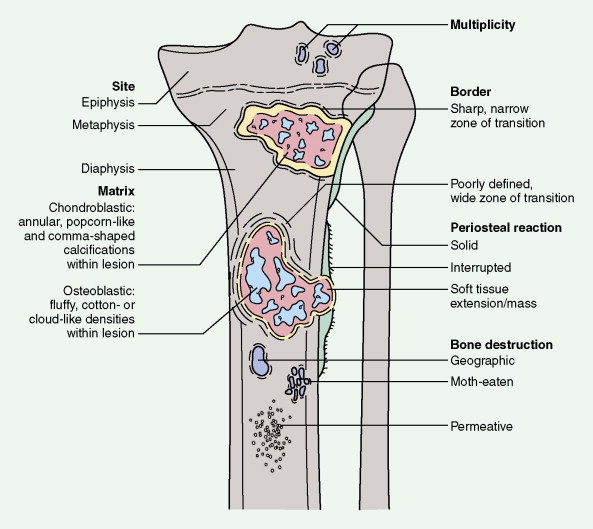
As with any soft tissue mass, tumors of bone may also be benign or malignant. Primary malignant lesions and secondary malignancies (i.e., those arising in association with a prior benign tumor) must be distinguished from metastatic lesions (which are much more common). Each of the structural components of bone may give rise to benign or malignant neoplasms, and it is important to recognize that certain benign lesions have the potential for malignant transformation. Radiographs of a lesion can help in the assessment of whether a lesion is benign or malignant, and in some cases such imaging studies can even suggest the histopathologic subtype of the bone tumor.
Osteosarcoma
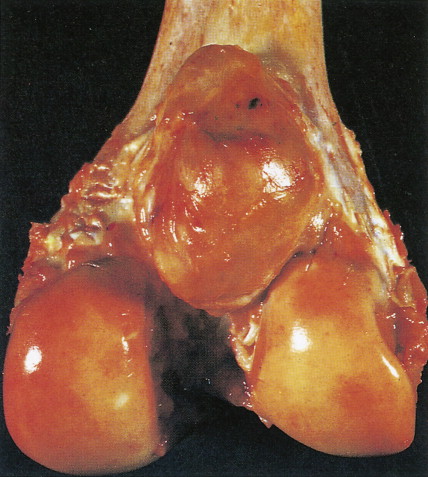
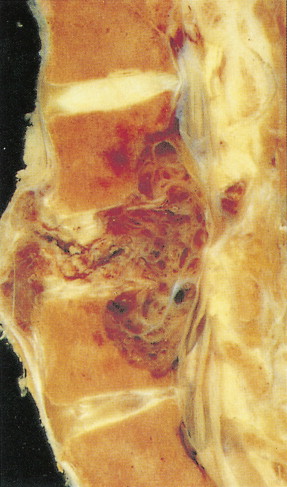
Defined as an osteoid-producing primary malignancy of bone, osteosarcoma is the most common primary sarcoma of bone and the second most common primary bone tumor after plasma cell myeloma. It develops in men slightly more frequently than in women, with an incidence ratio of 1.5:1. The distribution by age is bimodal. During adolescence, osteosarcoma arises in areas of rapid growth, for example, around the epiphyses of long bones. In some studies adolescents with this tumor have been found to be taller than age-matched controls. In the second peak in incidence, among older patients, osteosarcoma develops most frequently in areas of prior benign bone lesions or in previously irradiated sites. For example, solitary lesions of osteochondroma rarely give rise to osteosarcoma, but in patients with multiple lesions (enchondromatosis or Ollier disease) osteosarcoma develops more frequently. In 0.2% of patients with Paget’s disease, osteosarcoma arises in pagetoid bone. Multicentric tumors have developed in patients with prior chronic radium ingestion, such as watch-dial painters, and occasionally in the absence of any known risk factors, usually in children under age 10. Patients with familial retinoblastoma (i.e., those who have inherited deficiencies of the RB tumor suppressor gene) have a greatly increased risk of developing osteosarcoma (nearly a 10% lifetime risk, hundreds of times higher than the risk in the general population).
Patients often present with symptoms of bone pain, which may be indolent or of relatively short duration. Alkaline phosphatase levels are generally elevated, except in undifferentiated osteosarcoma, and they have prognostic value; elevated values after amputation herald residual or relapsing disease. (It should be remembered that normal values in children are higher than in adults.)
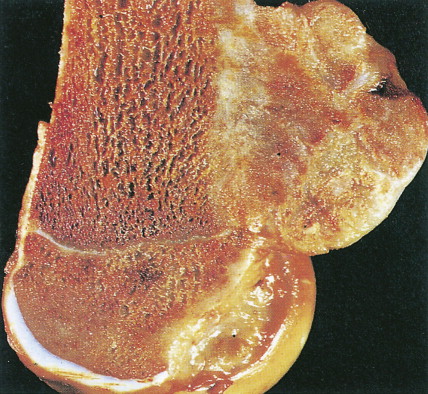
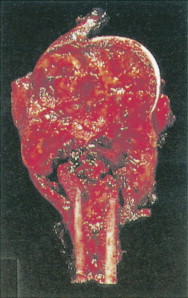
Characteristic radiographs and computed tomography (CT) scans of primary lesions reveal osteolysis and periosteal new bone formation; later-stage tumors may show cortical destruction and breakthrough into soft tissues. The telangiectatic variant of osteosarcoma is almost entirely lytic. Periosteal elevation results in the classic “Codman triangle” on radiography. Ossification may be slight, moderate, or densely sclerotic. Another pattern of periosteal reaction can be identified by a “sunburst” appearance of ossification. This phenomenon results from newly formed bone spicules oriented at right angles to the cortical soft tissue extension. (In the gross specimens shown among the figures that follow, it is instructive to note the morphology that results in the characteristic radiographic appearance of osteosarcoma.) In addition to plain radiography, CT examination, including scans of the chest, is essential to evaluate the stage of a lesion; CT scans of the chest help detect pulmonary metastases, the most likely site of disseminated disease. Radionuclide bone scan imaging is useful in the staging evaluation of osteosarcoma. This modality generally reveals intense uptake within the lesion, and may detect skip lesions, metastases, or a multicentric primary tumor. Uptake in the lungs on bone scan may identify early pulmonary lesions. 2-[ 18 F]-fluoro-2-deoxy- d -glucose–positron emission tomography (FDG-PET) scans are being increasingly evaluated as a tool to assess both extent of disease and, possibly, response to therapy.
The demonstration of “malignant osteoid” (i.e., noncalcified bony substance produced by the tumor cells themselves) is required for a histologic diagnosis of osteosarcoma. Based on the predominant cell type, osteosarcomas can be classified into osteoblastic (45% of cases), chondroblastic (27%), anaplastic (17%), fibroblastic (9%), and telangiectatic (1%) variants. Although most reports have shown that histologic subclassification has little prognostic value, histologic grade seems to correlate with tumor behavior.
Juxtacortical (parosteal) osteosarcomas are relatively uncommon (3% to 4%), often low-grade variants that arise equally in either sex. Patients with these tumors are about a decade older than patients with conventional higher-grade types. Gross examination shows bulky tumors that tend to encircle the cortex of bone, generally the distal femur, and less commonly the proximal humerus. The radiologic differential diagnosis includes osteochondroma and myositis ossificans.
Multimodality treatment is the standard approach to management of localized osteosarcoma. Preoperative (“neoadjuvant”) and/or postoperative (“adjuvant”) chemotherapy for osteosarcoma is now accepted as standard treatment. Histologic evidence of tumor necrosis following preoperative chemotherapy is among the most powerful prognostic factors for survival of patients with osteosarcoma. Doxorubicin and cisplatin are the core agents of adjuvant regimens in osteosarcoma, but ifosfamide and methotrexate may also have useful roles. Although these aggressive combination-chemotherapy regimens can be associated with considerable toxicity, overall survival is significantly improved with chemotherapy when compared with surgical resection alone.
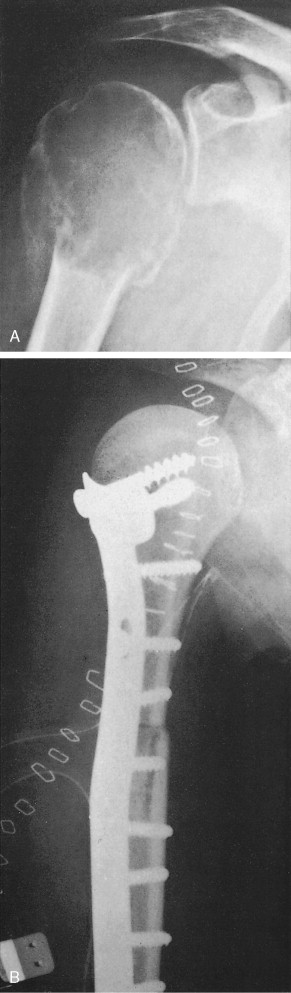
Besides the routine use of chemotherapy in osteosarcoma management, function-sparing surgery has been shown to be equivalent in overall survival when compared to more debilitating amputations, which were the standard of care before the 1970s. Current treatment approaches combine chemotherapy with local resection rather than amputation, with encouraging results in terms of survival rates and the functional status of affected limbs. Intensive chemotherapy with surgical intervention should be considered if metastases develop in the lung. Some patients who present initially with radiographically evident limited metastatic disease in the lung may be cured by resection of both the primary lesion and the pulmonary metastases, combined with systemic chemotherapy.
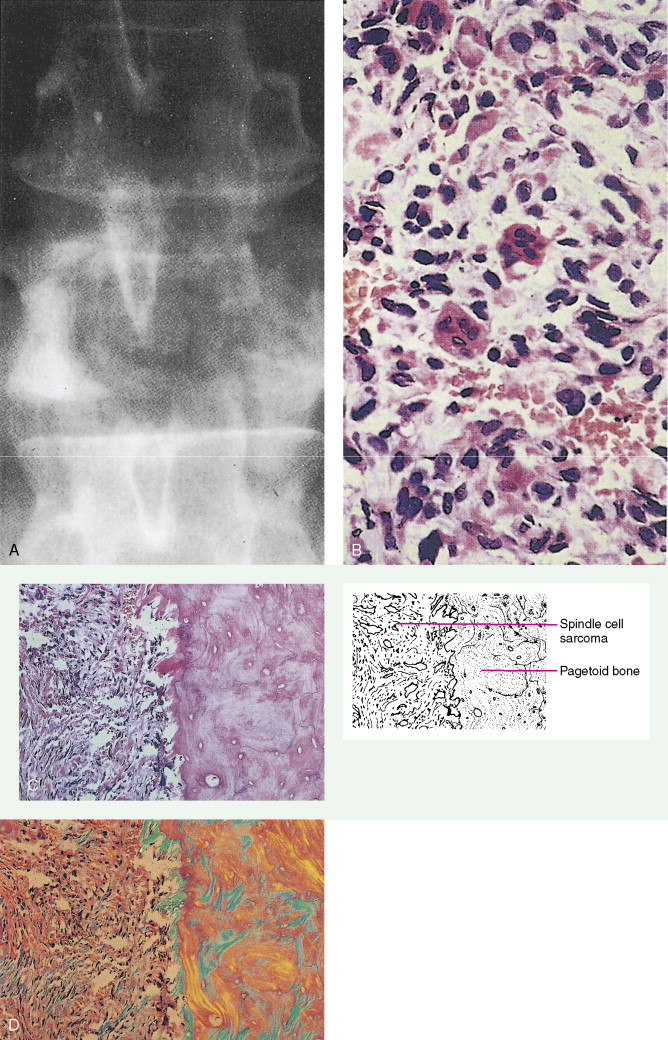
In sites of prior Paget’s disease or irradiation, osteosarcoma seems to be responsive to the usual therapeutic methods. However, because of its more central location and the vascularity of pagetoid bone, curative resection tends to be difficult.
Ewing Sarcoma
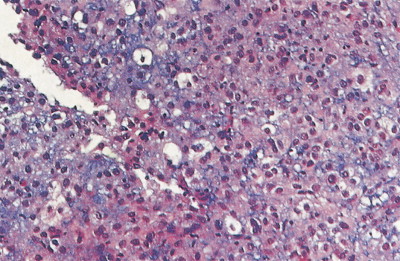
Ewing sarcoma is a high-grade small round cell sarcoma of unknown cellular origin. Ewing sarcoma was once considered primarily a bone tumor, but it is increasingly recognized as a primary malignancy of soft tissues as well. Cytogenetically, it is usually characterized by chromosomal translocations involving chromosome 22 (most often an 11;22 translocation, identical to that found in primitive neuroectodermal tumors). The molecular identity of Ewing sarcoma with primitive neuroectodermal tumors has led to a grouping known as the Ewing sarcoma family of tumors based on the molecular pathology; Ewing sarcoma and primitive neuroectodermal tumor are now considered synonymous. Ewing sarcoma accounts for about 10% to 14% of primary malignant bone tumors in whites, but it is rare in blacks. Its incidence peaks between the ages of 10 and 25 years, with a 2:1 male-to-female ratio. Both Ewing sarcoma and osteosarcoma occur primarily in the same sex and age group, but they can be clinically distinguished by other characteristics. The most common sites of occurrence include the femur (27%), pelvis (18%), and tibia or fibula (17%).
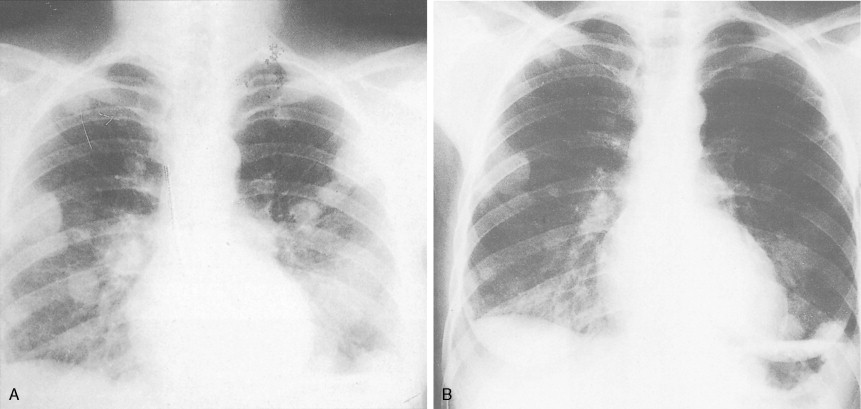
Patients with Ewing sarcoma of bone often present with pain and a rapidly enlarging mass and may have fever, leukocytosis, and an elevated erythrocyte sedimentation rate (sometimes mimicking osteomyelitis). Early diagnosis of Ewing sarcoma involving the pelvic bones may be delayed because of poorly localized pain and a clinically inapparent mass. Patients may also have pulmonary symptoms, indicating lung metastases, or spinal cord compression, resulting from metastatic deposits in vertebrae. Radiographically, Ewing sarcoma characteristically presents as a fusiform enlargement of the long bones with central mottling (“cracked ice”), indicating a permeative type of bone destruction, and “onion-skin” layering of the periosteal reaction. Initial diagnostic evaluation should include CT and/or magnetic resonance imaging (MRI) scans of the primary site, as well as imaging of the chest and liver, since those are common sites of disease spread. Additionally, a baseline bone scan should be obtained to rule out occult metastases at diagnosis. Ewing sarcoma can be imaged by FDG-PET scans as well as by labeled octreotide scans, although the value of these more sophisticated imaging studies requires further clarification.
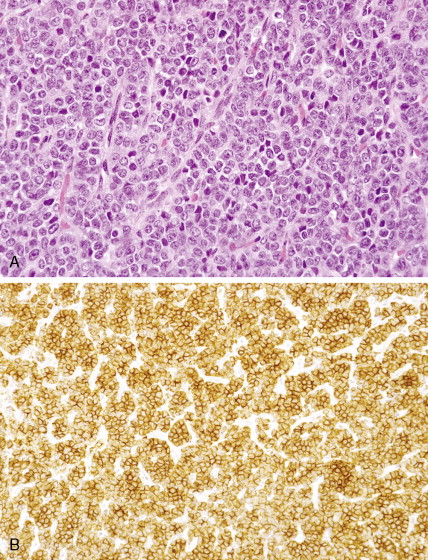
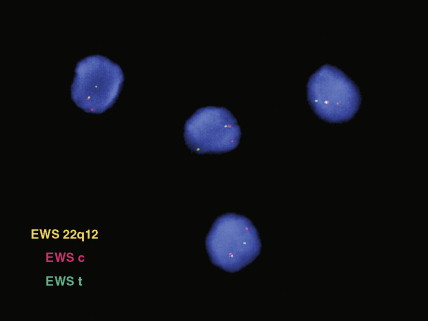
Microscopic examination of biopsy specimens typically reveals cytologically uniform, small, round cells with scant cytoplasm arranged in sheets, bordered by fibrous septae. Immunohistochemical staining for CD99 (O-13) reveals diffuse membranous reactivity. The periodic acid–Schiff stain is positive for glycogen, which can be digested by the diastase reaction. Electron microscopy can confirm the presence of large quantities of glycogen. The latter two features have largely been supplanted by immunohistochemistry. Cytogenetic evaluation, looking for translocations involving the long arm of chromosome 22 (most commonly t(11;22)), or fluorescence in situ hybridization (FISH) evaluation for EWSR1 rearrangement, is helpful to confirm the diagnosis, particularly in small biopsy specimens or when the histologic and/or clinical features are not typical. FISH can be performed on paraffin-embedded archival material.
Current aggressive multimodality treatment results in long-term disease-free survival in about 70% of children under 16 years of age who present with localized Ewing sarcoma. It is estimated that up to 10% of patients with metastases may also be cured, although this is generally noted in the pediatric population. Metastases most frequently involve the lung, bone, bone marrow, and spinal cord. Survival rates correlate inversely with age: 70% of patients under 10 years of age survive as compared with 46% of those 16 years and older. This may be due in part to differences in the resectability of the disease and a disproportionate finding of unresectable pelvic and proximal sites of disease in adults, or perhaps due to a difference in the ability of older adults to tolerate the high-dose chemotherapy required for adequate treatment.
Chondrosarcomas
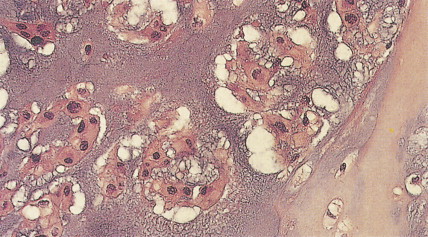
Malignant tumors of bone that produce cartilage but no osteoid are defined as chondrosarcomas. They account for 17% to 22% of primary malignant bone tumors and are the second most common bone sarcoma. Their incidence increases steadily with age. Primary lesions occur in previously normal bone, whereas secondary chondrosarcomas arise in prior benign lesions, most frequently enchondromas. Malignant degeneration in multiple enchondromatosis (Ollier disease) is reported in patients with this condition alone or in association with soft tissue hemangiomas (Maffucci disease). Chondrosarcomas in patients with multiple enchondromatosis are generally low-grade. About a tenth of radiation-associated sarcomas are chondrosarcomas, and secondary tumors may also arise in bone affected by Paget’s disease. The most common sites of involvement for primary chondrosarcoma are the pelvis (31%), femur (21%), shoulder (13%), ribs (9%), and face (9%). Lesions may be painful, especially if they increase rapidly in size, or they may present as a painless mass.
Radiographically, central chondrosarcomas show fluffy, popcorn-like calcifications. Peripheral tumors, on the other hand, tend to have long, lightly calcified spicules radiating from the cortex to a flattened outer surface, with little evidence of cortical or medullary involvement. A faint Codman triangle may be evident as a result of lifting of the periosteum. Chondrosarcomas tend to take up radionuclides avidly during bone scanning. In their gross aspect, chondrosarcomas have translucent, bluish white, mucoid surfaces; reactive new bone formation is seen in slow-growing lesions, or cortical destruction may be evident, marking high-grade, rapidly enlarging lesions. Its microscopic appearance is characterized by the appearance of cartilage with malignant chondrocytes. Especially in low-grade lesions, the histologic character of a tumor is an unreliable predictor of biologic behavior, since similar lesions may behave differently depending on age, site of origin, lesion size, and particularly radiographic appearance. Rare variants include clear cell chondrosarcoma and mesenchymal chondrosarcoma. The clear cell type, which occurs predominantly in the femoral head or humeral head of adult men, is marked by local recurrences and eventual metastases. Mesenchymal chondrosarcoma is characterized histologically by a small round cell tumor with foci of well-differentiated cartilage. This tumor tends to arise in the ribs, mandible, maxilla, skull, and extraskeletal sites. Extraskeletal myxoid chondrosarcoma (which nearly always arises in deep soft tissue and very rarely in bone), despite its name, is not a chondrosarcoma at all but instead a distinctive myxoid sarcoma of uncertain lineage characterized by translocations involving the EWS gene on chromosome 22q.
Because local recurrences generally increase the histopathologic grade of the tumor, and because chondrosarcomas are quite resistant to both radiation therapy and chemotherapy, every effort should be made to resect the primary lesion completely. Thus, expert aggressive resection is indicated, particularly of eminently curable, low-grade chondrosarcomas.
Giant Cell Tumor of Bone
Constituting 5% of primary tumors of bone, giant cell tumor of bone is an unusual lesion that may behave locally in an aggressive manner but has a very low metastatic potential. Incidence of the lesion peaks in the third decade. Fifty-five percent of affected individuals are women. Half of giant cell tumors of bone are located around the knee, arising in the distal femur, patella, or proximal tibia or fibula. Lesions may rarely be associated with areas of active Paget’s disease.
Radiographically, giant cell tumors are generally eccentrically situated, epiphyseal lesions having a central lucency and increasing density toward the periphery. No new bone formation is present in actively growing lesions. Grossly, the tumor may appear solid despite a cystic appearance on radiography. Microscopically, it is marked by prominent, multinucleated osteoclast-like giant cells dispersed throughout well-vascularized stromal tumor tissue containing numerous mononuclear cells. The mononuclear cells have nuclei identical to those of the giant cells. Mitotic figures may be present and have no prognostic significance. Although fewer than 10% of these tumors are malignant on first presentation, up to 30% assume a malignant behavior after multiple recurrences.
The prognosis of giant cell tumors is difficult to predict. A recurrence rate of 50% or more has been reported after curettage; most patients require multiple therapeutic interventions before disease is successfully eradicated. Wound implantation may result in recurrences in soft tissue. Sarcomatous transformation is rare and may be related to prior radiation therapy. Case reports of putatively “antiangiogenic” therapy being useful in the management of metastatic, unresectable giant cell tumor of bone suggest that this tumor type may be sensitive to agents such as recombinant human interferon-α, but these anecdotes have not been confirmed in larger trials. The most interesting approach to the management of giant cell tumor of bone involves the monoclonal antibody known as denosuman, which targets the osteoprotegerin/receptor activator of nuclear factor κB (RANK) ligand.
Chordoma
Chordoma, representing 1% of malignant bone tumors, arises from remnants of the embryologic notochord, generally in the cervical or sacral area. The peak incidence is in the fifth to seventh decades. Sacral lesions generally occur in older patients, with a median age of 56 years as compared with 47 years for cervical lesions. Sacral lesions result in pain, usually of over a year’s duration before diagnosis; the pain is sometimes referred to the hip or knee. Neurologic bowel or bladder symptoms may be the presenting manifestations. Eighty percent of patients with cranial chordomas report headache. Cranial nerve signs, chiasmal involvement, or endocrine abnormalities may be present. Nasopharyngeal chordomas present as a mass with nasal obstruction before neurologic involvement. Osteolysis of one or more vertebral bodies and a soft tissue mass herald the presence of a sacral chordoma. Cranial chordomas present with destruction of the sella, the clinoid, the clivus, and the apices of the petrous bones. On gross examination, chordomas are lobulated, pseudoencapsulated masses varying in consistency from jelly-like to cartilaginous. Microscopically, the tumor is composed of cords of epithelioid cells with abundant eosinophilic cytoplasm. Intracellular and extracellular mucin production is prominent, and distinctive physaliferous (“bubble”) cells are often present. Mitotic figures are uncommon.
Treatment consists of resection and radiation therapy, including promising data using newer radiotherapeutic techniques such as highly focused radiation therapy via proton beam accelerators. Systemic therapy testing the use of tyrosine kinase inhibitors (TKIs) is also being evaluated for unresectable or metastatic disease. Complete resection is seldom possible. Local failure is common, and 5% to 43% of these tumors eventually metastasize. The median survival is about 6 years.
| Benign Lesion | Resultant Malignancy |
|---|---|
| Enchondroma: in long or flat bones; in short tubular bones only in Maffucci syndrome or Ollier disease | Chondrosarcoma |
| Paget’s disease | Osteosarcoma |
| Unclassified sarcoma (rare) | |
| Neurofibroma: in plexiform neurofibromatosis | Malignant schwannoma |


| Feature | Osteosarcoma | Ewing Sarcoma |
|---|---|---|
| Site of involvement | ||
| Long bone | Metaphysis | Diaphysis |
| Flat bone | Rare | Common |
| Medullary cavity | Rare | Common (moth-eaten appearance) |
| New bone formation | Common | Only secondary |
| Periosteal reaction | Codman triangle spiculation (sunburst) | Lamellated “onion-skinning” |
| Soft tissue mass | Less prominent | Large, common |
| Radiation-associated | Yes | No |
| Precursor lesions | Yes | No |
SARCOMAS OF SOFT TISSUES
Soft tissue sarcomas are a widely heterogeneous family of tumors found in all age groups; they range from orbital rhabdomyosarcoma, which peaks in incidence at 4 years, to liposarcoma, the most common sarcoma in adults over age 50. Forty percent of soft tissue sarcomas develop in the lower extremities, most commonly in the thigh; these tumors may also arise in virtually any site of the body, such as the trunk, head or neck, arm, and retroperitoneum. Patients usually present with a solitary, painless, palpable mass on the extremities or trunk. Intra-abdominal and retroperitoneal primary tumors, when advanced and large, may cause symptoms related to invasion or displacement of organs, weight loss, and pain. The duration of symptoms ranges from a few weeks to decades, with a median of 1–3 months. Soft tissue sarcomas have developed within ports of prior radiation therapy delivered 2–20 years earlier (median about 10 years).
Preoperative evaluation requires CT or MRI scan of the primary lesion to assess the anatomic extent of disease involvement locally. If the lesion abuts bone, MRI may be particularly helpful to determine whether there is periosteal invasion or reaction. Uptake of radionuclide on bone scan is usually not documentation of involvement by tumor but rather indication of a reaction; however, if this is documented at a site distant from the primary, the presence of occult bony metastases should be considered in sarcomas that have a predilection to spread to the bones, such as myxoid liposarcoma, endometrial stromal sarcoma, and angiosarcoma. Staging is directed at determining whether distant spread of disease has occurred, and this should be performed before starting definitive local therapy. Staging generally requires a CT scan of lungs and liver, the two most common sites of metastasis for soft tissue sarcomas.
The gross appearance of most soft tissue sarcomas is not particularly discriminatory or distinctive. The histopathology, however, varies greatly with the specific subtype of sarcoma, and these distinctions can be subtle. There is no substitute for having pathology reviewed by a pathologist with specialty expertise in the diagnosis of soft tissue sarcomas. Grossly, soft tissue sarcomas are often pseudoencapsulated, fleshy tumors that grow along anatomic tissue planes, thus requiring wide excision beyond the apparent border of the grossly evident disease for local control. However, most soft tissue sarcomas are relatively circumscribed and push aside adjacent structures. Some tumors are characterized by invasion of adjacent organs and prominent areas of necrosis, particularly centrally, as the tumor outgrows its blood supply. Because of microscopic projections of tumor beyond the apparent “capsule,” local recurrence follows a “shelling out” procedure in about 80% of cases. Therefore, it is critical to consider a proper surgical repeat resection when a margin-positive “shelling out” surgical procedure had previously been performed.
A summary of the American Joint Cancer Committee (AJCC) staging system for soft tissue sarcomas is shown in Table 12.5 . Tumor stage, which is determined by both grade and size, correlates with survival. The 5-year survival rate for soft tissue sarcomas arising in various anatomic locations is similar when it is adjusted for grade. The exceptions to this pattern are intra-abdominal and retroperitoneal primary tumors; these lesions, even if low grade, tend to be large, and at the time of diagnosis they often involve vital organs.
| Regional Lymph Nodes (N) | |
|---|---|
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastasis (M) | |
|---|---|
| MX | Distant metastasis cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| Stage Grouping | ||||||
|---|---|---|---|---|---|---|
| 1 | T1a | N0 | M0 | G1–2 | G1 | Low |
| T1b | N0 | M0 | G1–2 | G1 | Low | |
| T2a | N0 | M0 | G1–2 | G1 | Low | |
| T2b | N0 | M0 | G1–2 | G1 | Low | |
| II | T1a | N0 | M0 | G3–4 | G2–3 | High |
| T1b | N0 | M0 | G3–4 | G2–3 | High | |
| T2a | N0 | M0 | G3–4 | G2–3 | High | |
| III | T2b | N0 | M0 | G3–4 | G2–3 | High |
| IV | Any T | N1 | M0 | Any G | Any G | High or low |
| Any T | N0 | M1 | Any G | Any G | High or low | |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


