Hodgkin Disease
About 7400 new cases of Hodgkin disease are diagnosed in the United States each year, with a slightly higher incidence in men. A bimodal age distribution is observed in the Western world, with peaks at 30 and 70 years of age. In Japan, however, this disease is less common in young adults. A bimodal distribution is not seen in underdeveloped countries, suggesting that Hodgkin disease is an uncommon, late complication of a common childhood infection. However, the cause is unknown.
CLASSIFICATION AND IMMUNOBIOLOGY
The histologic features of Hodgkin disease vary widely and may not exhibit the classic criteria for malignancy. Moreover, in about 10% to 15% of cases, recognition of the disorder and its distinction from benign and other malignant lymphoproliferative disorders is difficult. In these cases surface marker studies may be extremely important, and accurate diagnosis may depend on the availability of fresh tissue for evaluation by an experienced hematopathologist.
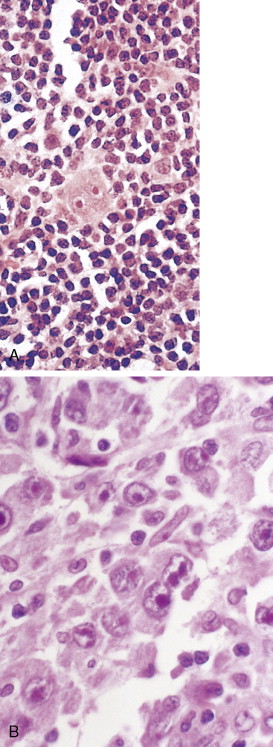
The first histopathologic classification of Hodgkin disease was established in 1966 by an international conference in Rye, New York, and consists of four subtypes. The subtypes are still included in the new World Health Organization (WHO) classification (see Fig. 16.1 ). The majority of cells composing a tumor mass appear to be benign or reactive and include lymphocytes, eosinophils, granulocytes, plasma cells, fibroblasts, and mononuclear cells. The pathognomonic finding for the diagnosis is the Reed-Sternberg cell in the appropriate immunoreactive background.

Molecular biologic analysis of micromanipulated Reed-Sternberg cells supports the B lymphocyte as the cell of origin. It is noteworthy that cells morphologically indistinguishable from Reed-Sternberg cells have been found in other hematologic disorders, both benign (e.g., infectious mononucleosis) and malignant (e.g., angioimmunoblastic T-cell lymphoma). Surface marker studies are useful in the differential diagnosis. The immunophenotype of the typical Reed-Sternberg cell is positive for Ki-1 (CD30), CD15 (Leu-M1), HLA-DR, and CD25 (interleukin-2 [IL-2] receptor); it is negative for leukocyte common antigen (LCA) (CD45) and is usually negative for T-cell and B-cell antigens, as well for most monocyte, macrophage, and histiocyte antigens. An exception is lymphocyte-predominant Hodgkin disease in which Reed-Sternberg cells have the immunophenotype of B cells.
The Reed-Sternberg cell is known to secrete a number of cytokines, including interleukin-1 (IL-1), IL-5, IL-6, IL-9, tumor necrosis factor-α, macrophage colony-stimulating factor, and transforming growth factor-β. These cytokines may attract the mixed inflammatory cell infiltrate seen in Hodgkin disease, result in the characteristic fibrosis seen in the nodular sclerosis subtype, and contribute to the immunosuppression that may occur in Hodgkin disease. Epstein-Barr virus (EBV) has been associated with a significant percentage of cases of Hodgkin disease, particularly the mixed-cellularity type, by a number of investigational methods.
CLINICAL EVALUATION AND STAGING
Evaluation of a patient includes a detailed history and physical examination, complete blood cell count with differential, blood chemistries, chest radiograph, computed tomography (CT) scan of the chest, abdomen, and pelvis, and bone marrow aspiration and biopsy. Historically, gallium-67 citrate scanning, using a dose of 10 nCi, with delayed views up to 72 and 96 hours if necessary, could reveal clinically inapparent sites. In addition, gallium scans were useful in the differential diagnosis of residual masses and in following the response to therapy. Increasingly, gallium scanning has been replaced by positron emission tomography (PET). PET is more sensitive than gallium scanning, and the whole study can be accomplished in a matter of hours without the inconvenience of bringing the patient back to the radiology suite for delayed images on subsequent days. As with gallium scans, PET may be useful in evaluating residual masses after chemotherapy, with residual activity on PET scan suggesting residual disease. Staging laparotomy and lymphangiography are of historical interest but have been rendered obsolete by modern imaging techniques and the routine use of chemotherapy as part of the treatment regimen.
In most cases, Hodgkin disease tends to progress in an orderly fashion from one lymph node–bearing area to the next. The results of the staging workup are a part of the Ann Arbor staging system ( Fig. 16.13 ). The mainstay of treatment is chemotherapy. Historically, many patients were treated with radiation therapy alone, but outcomes are generally better when chemotherapy is added. Only rare patients with limited stage I disease or patients who are too elderly or infirm to tolerate chemotherapy are treated with radiation alone. Whether or not radiation is added to chemotherapy depends on the stage of disease, the bulk of disease, and the chemotherapy regimen chosen, among other factors.

CLINICAL MANIFESTATIONS
Most patients present with localized disease. Lymph node enlargement may wax and wane for unknown reasons, although “spontaneous” remissions are rare. About 20% to 30% of cases have B symptoms (fever of >38°C, unexplained weight loss of >10% of body weight in 6 months, and/or night sweats); however, only 5% to 10% of patients present with extranodal sites of disease, including lung, liver, and bone marrow. The latter presentation is more often seen in patients over 60 years of age.

Of note, nodular lymphocyte-predominant Hodgkin disease seems to behave differently than classical Hodgkin disease and more like an indolent lymphoma. For instance, late relapses, even 20 or more years after therapy, and subsequent development of non-Hodgkin lymphoma are much more common with nodular lymphocyte-predominant Hodgkin disease than with classical Hodgkin disease. Whether nodular lymphocyte-predominant Hodgkin disease should be treated differently than classical Hodgkin disease remains a source of controversy.














Non-Hodgkin Lymphoma
Non-Hodgkin lymphomas (NHLs) are a diverse group of malignancies of the lymphoreticular system that have heterogeneous histopathologic, immunologic, cytogenetic, and clinical characteristics. About 55,000 new cases are diagnosed in the United States each year, and the incidence is rising. Although the disease can appear at any age, the median age is 67 years and the incidence increases with age. Indolent NHLs are particularly rare in children. NHL is slightly more prevalent in men than in women. Although the etiology is unknown, malignant lymphomas are more common in patients with immune deficiency syndromes (e.g., acquired hypogammaglobulinemia), autoimmune disorders (e.g., rheumatoid arthritis), immunosuppressive states (e.g., renal transplants), chronic hepatitis B and C, and AIDS. In AIDS patients, for example, 5% to 10% develop intermediate- to high-grade B-cell NHLs, and these are included as part of the Centers for Disease Control and Prevention (CDC) AIDS diagnosis criteria. EBV has been observed in a significant percentage of AIDS-associated lymphomas and seems to have an etiologic role in their pathogenesis (see Chapter 19 ). The African form of Burkitt lymphoma is associated with EBV infection as well. A form of aggressive T-cell leukemia-lymphoma associated with human T-cell lymphoma-leukemia virus (HTLV-1) infection has been reported in southern Japan, the Caribbean, and the southeastern United States. Genetic factors may also predispose to lymphomas, as evidenced by cases in patients with chromosomal disorders (e.g., ataxia-telangiectasia) and those with immunodeficiency disorders other than AIDS.
CLASSIFICATION AND IMMUNOBIOLOGY
Histopathologic classifications and frequency of NHL subsets are shown in Figure 16.35 and Tables 16.1 and 16.8 . The “working formulation” classification (see Fig. 16.35 ), devised by an international panel of clinicians and pathologists sponsored by the National Cancer Institute (NCI), retained the relevant features of the previous Rappaport system and divided lymphomas into 10 separate subgroups (A to J) and three major prognostic categories. These classification schemes have been replaced by the WHO classification of lymphoid neoplasms based on morphologic, immunophenotypic, and cytogenetic features ( ). Broadly the WHO classification identifies two subsets of lymphoid neoplasms: B-cell and T/NK (natural killer)-cell neoplasms. The B and T/NK subgroups are then further subclassified as precursor or mature neoplasms. Clinically, the histologies can be considered in three broad categories—indolent, aggressive, and highly aggressive—based upon the natural history of the disease.

| Category | Subgroup | Frequency (%) † | Survival Range (yr) |
|---|---|---|---|
| Low grade | A | 14 | |
| B | 26 | 5–10 | |
| C | 19 | ||
| Intermediate grade | D | 4 | |
| E | 8 | ||
| F | 7 | 2–5 | |
| G | 22 | ||
| High grade | H | 9 | |
| I | 5 | 0.5–2 | |
| J | 6 |
* See Figure 16.35 for definitions. Working Formulation subgroups G and H are often called diffuse large cell lymphoma (DLCL) and treated as high-grade or poor-prognosis lymphomas. “Aggressive” lymphomas refer to subgroups E to H. “Very aggressive” lymphomas refer to subgroups I and J.
A considerable understanding of the immunology of lymphomas has been achieved by the availability of specific monoclonal antibodies (MAbs) directed against cell surface antigens (see Figs. 16.37 and 16.38 and Table 16.5 ). It has been found that the malignant cells of most lymphomas have normal cell counterparts. By use of an extensive panel of MAbs, about 90% of lymphomas can be identified as B-cell lymphomas and 10% as T-cell lymphomas. With modern immunohistochemical and genetic techniques, what were once thought to be true “histiocytic” lymphomas are now recognized as B- or T-cell neoplasms in most cases or true histiocytic disorders such as histiocytic sarcoma in others.


| Neoplasm | CD19 | CD20 | CD22 | CD10 | CD5 | CD25 | CD11c | CD21 | CD38 | TdT |
|---|---|---|---|---|---|---|---|---|---|---|
| Common ALL | + | ± | ± | + | − | − | − | − | +/− | + |
| Small non–cleaved cell Burkitt lymphoma | + | + | + | + | − | − | − | +/− | − | − |
| Large cell/large cell immunoblastic lymphoma | + | + | + | ± | − | ± | − | ± | + | − |
| Nodular (follicular) lymphomas | + | + | + | + | − | − | − | + | +/− | − |
| Mantle cell lymphoma | + | + | + | −/+ | + | − | − | +/− | − | − |
| Marginal zone lymphoma | + | + | + | − | − | +/− | +/− | − | +/− | − |
| Small lymphocytic/CLL | + | + | + | − | + | − | − | +/− | − | − |
| Small lymphocytic-plasmacytoid (Waldenstrom’s macroglobulinemia) | +/− | +/− | +/− | − | +/− | + | − | − | + | − |
| Hairy cell leukemia | + | + | + | − | − | + | + | − | −/+ | − |
| Myeloma | − | − | − | − | − | − | − | − | + | − |
* Cluster designation (CD) groupings are noted as well as the commonly used terms.
Most lymphomas express LCA (CD45), which is detectable by immunoperoxidase staining using MAbs specific to LCA; LCA staining is very useful because it is immunoreactive (positive) even in poorly differentiated lymphomas that might otherwise be mistaken for carcinoma, sarcoma, or melanoma, all of which are LCA-negative. Furthermore, LCA staining can be performed on formalin- and B5-fixed tissues. (The latter is preferred because of better histologic detail, particularly of the cell nucleus.) Cell lineage as determined by the use of MAbs usually requires fresh tissue. The monoclonality of B-cell lymphomas can be confirmed by immunoperoxidase staining for one class of immunoglobulin light chain or by immunoglobulin gene rearrangement studies. T-cell lymphomas, on the other hand, do not express cell surface immunoglobulins, but many show clonal loss of expression of pan–T-cell antigens, which can be tested using MAbs. Gene rearrangement studies of the T-cell receptor may also be used to determine monoclonality, although lack of monoclonality does not exclude the diagnosis of lymphoma. Lymphomas of T-cell lineage can also be identified by the formation of so-called “rosettes” on reaction with sheep erythrocytes. T-cell phenotyping is carried out by reaction with available MAbs (see Fig. 16.38 ).
Many chromosomal, molecular, and genetic defects have been detected in various lymphomas. These are described in the section on lymphoma subsets (see below). A better understanding of the biology of lymphomas allows for clarification of the pathogenesis and also for future novel therapeutic approaches.
Amplification of DNA can now be performed on small amounts of tissue by the polymerase chain reaction technique, thus allowing for detection of abnormal clones (i.e., monoclonal population of malignant cells). The method is extremely sensitive and may detect residual tumor or early disease relapse.
Histologic Subtypes
Immunophenotypic and other classification markers are noted in Figures 16.37 to 16.40 and Table 16.5 .


B-Cell Non-Hodgkin Lymphomas
B-CELL CHRONIC LYMPHOCYTIC LEUKEMIA/SMALL LYMPHOCYTIC LYMPHOMA
This subtype of NHL is a generally indolent lymphoma with a favorable prognosis. The tumor is characterized by a diffuse pattern of growth with effacement of nodal architecture by round lymphocytes of small to medium size. Mitotic figures are rare. In a minority of cases there may be plasmacytic differentiation with associated macroglobulinemia. Small lymphocytic lymphoma (SLL) and chronic lymphocytic leukemia (CLL) probably represent different clinical manifestations of the same disease process, the former marked mainly by adenopathy and the latter by lymphocytosis involving the bone marrow and peripheral blood.
Until recently, identifying cytogenetic abnormalities in SLL/CLL cells was difficult. With modern cytogenetic techniques and fluorescence in situ hybridization, however, chromosomal abnormalities can be detected in 70% to 80% of SLL/CLL cells ( ). These abnormalities can give insight into the pathogenesis of the disease and are also useful in predicting clinical behavior. Deletion of 17p and 11q seem to predict more aggressive disease, whereas deletion of 13q predicts a more indolent course. Normal cytogenetics and trisomy 12 predict an intermediate course. Other factors, such as increased expression of CD38, increased expression of ZAP-70 ( ), deletion of the p53 gene, and an unmutated immunoglobulin heavy-chain gene also can predict a more aggressive clinical course ( ). None of these markers, however, is perfect, and the interplay of the various prognostic markers is complex. Thus, the clinical management of the patient is still dictated by the behavior of the disease and the presence or absence of disease-related side effects. FLOAT NOT FOUND
LYMPHOPLASMACYTIC LYMPHOMA
Lymphoplasmacytic lymphoma (LPL) is a rare B-cell neoplasm composed of a diffuse proliferation of small B lymphocytes, plasmacytoid lymphocytes, and plasma cells. Frequently the disease is associated with an IgM monoclonal gammopathy. The combination of lymphoplasmacytic lymphoma with an IgM monoclonal gammopathy is termed Waldenstrom’s macroglobulinemia. Accumulation of IgM in the cytoplasm of LPL cells results in characteristic Dutcher bodies. LPL is generally an indolent disease, although high levels of IgM can cause hyperviscosity syndrome resulting in headache, epistaxis, visual changes, and coma necessitating plasmapheresis to remove the antibody and chemotherapy to debulk the disease and decrease antibody production.
MANTLE CELL LYMPHOMA
Mantle cell lymphoma is uncommon, accounting for only 3% to 4% of all cases of NHL. It is composed of small lymphocytes with intermediate-sized nuclei. The lymphoma appears to arise from B lymphocytes found in the mantle zone of lymphoid follicles. The histologic pattern of mantle cell lymphoma may be nodular, with neoplastic cells surrounding residual germinal centers (mantle zone pattern), or diffuse. Seventy percent or more of cases show a t(11;14) chromosomal translocation involving the BCL1 locus on chromosome 11, resulting in overexpression of the PRAD1 gene, the product of which, cyclin D1, is involved in cell cycle regulation. Also, over 70% of cases will show nuclear staining for cyclin D1. Expression of cyclin D1 can occur even in cases with no detectable t(11;14). Mantle cell lymphoma typically pursues an aggressive course with a median survival of 3–4 years, although rare cases do behave in an indolent fashion similar to CLL/SLL. The blastoid variant in particular can be very aggressive and can present with spontaneous splenic rupture. Mantle cell lymphoma has a predilection for the gastrointestinal (GI) tract with involvement in 15% to 30% of cases.
FOLLICULAR LYMPHOMA
Follicular lymphoma is the second most common B-cell NHL, accounting for approximately 35% of all cases. Follicular lymphoma is composed of both small centrocytes (cleaved follicle center cells) and large centroblasts (noncleaved follicle center cells). Follicular lymphoma is assigned a grade based upon the percentage of centroblasts present. Follicular lymphoma is classified as grade I when there are 5 or fewer centroblasts per high-power field (HPF), grade II when there are 6 to 15 centroblasts per HPF, and grade III when there are more than 15 centroblasts per HPF.
Most cases of follicular lymphoma exhibit a t(14;18) chromosomal translocation, which results in deregulation of expression of the BCL2 proto-oncogene on chromosome 18. This gene blocks programmed cell death (apoptosis), and its overexpression results in prolongation of the lifespan of involved cells, contributing to the pathogenesis of lymphoma. Of note, approximately 20% of diffuse large B-cell NHLs show the t(14;18) translocation, and therefore this translocation is not pathognomonic of follicular lymphoma.
MARGINAL ZONE LYMPHOMA (NODAL AND EXTRANODAL)
This low-grade B-cell NHL tends to involve lymph node sinuses and interfollicular zones and to surround follicles in a marginal zone pattern. Monocytoid B cells have small nuclei with irregular outlines and fairly abundant pale cytoplasm and may involve bone marrow and extranodal sites such as the salivary gland, with an indolent clinical course. The normal cellular counterpart of marginal zone lymphomas may be the marginal zone cells in the spleen. Though similar in appearance to hairy cell leukemia cells, marginal zone B-cell neoplasms are CD103-negative, whereas hairy cells are usually CD103-positive.
There are several important subtypes of marginal zone lymphoma. One is splenic marginal zone lymphoma with villous lymphocytes. Patients tend to present with left upper quadrant pain and early satiety from splenomegaly. Bone marrow involvement is common, but these patients tend to have comparatively little lymph node involvement. Splenectomy is often an effective palliative therapy and can result in regression of disease in other sites. Chemotherapy or immunotherapy is sometimes needed, although the disease tends to be indolent. Splenic marginal zone lymphoma with villous lymphocytes is associated with infection with viral hepatitis, and treating the hepatitis can lead to improvement in or resolution of the lymphoma.
Extranodal marginal zone lymphoma, also known as MALToma because it involves mucosa-associated lymphoid tissue, can occur at any number of mucosal sites and is notable for its association with certain underlying medical conditions. A common site of involvement is the stomach, where there is a strong association with Helicobacter pylori infection. This is discussed in more detail below. Extranodal marginal zone lymphomas of the eye have been associated with Sjögren syndrome and infection with certain strains of Chlamydia , whereas marginal zone lymphomas of the skin have been associated with infection with certain Borrelia species. Although there are reports of antibiotics being effective in treating marginal zone lymphomas occurring in these sites, the data are not as compelling as they are in gastric extranodal marginal zone lymphoma.
DIFFUSE LARGE B-CELL LYMPHOMA
Diffuse large B-cell lymphoma (DLBCL) is the most common NHL, accounting for approximately 40% of all cases of NHL. DLBCL follows an aggressive clinical course, and patients frequently present with rapidly enlarging lymphadenopathy and/or B symptoms such as fever, night sweats, and unintentional weight loss. The tumor is composed of large cells with prominent nucleoli. Notably, benign, reactive histiocytes are present in many cases of DLBCL.
There are several morphologic variants of DLBCL including the immunoblastic and centroblastic variants, although none of the variants have meaningful prognostic or treatment implications. T-cell/histiocyte-rich DLBCL can be easily confused with T-cell NHL or even Hodgkin disease. The distinction is important, since Hodgkin disease and T-cell NHL are treated much differently than DLBCL. This variant often presents in younger patients with disproportionate involvement of the bone marrow, spleen, and liver. At one time, T-cell/histiocyte-rich DLBCL was thought to have a worse prognosis than other DLBCLs, but when controlling for the International Prognostic Index (IPI) this no longer seems to be the case. The anaplastic variant of DLBCL can express CD30 and can also be easily confused with Hodgkin disease, especially since Hodgkin disease can express B-cell markers. Anaplastic DLBCL can also be confused with T/null anaplastic large cell lymphoma, another CD30 + neoplasm, although the two entities can easily be distinguished based upon CD20 staining and the lack of anaplastic lymphoma kinase (ALK) in anaplastic DLBCL.
Modern methods of treatment and the addition of MAbs to conventional chemotherapy have markedly improved the rate of survival of patients with these tumors, with approximately 50% to 60% of cases cured at present.
MEDIASTINAL DIFFUSE LARGE B-CELL LYMPHOMA
This variant in DLBCL presents with bulky mediastinal lymphadenopathy, often with relatively little disease in other areas. Mediastinal DLBCL tends to present in young patients with a slight female predominance and can mimic Hodgkin disease clinically and morphologically. Superior vena cava (SVC) syndrome is a common presentation. Although the disease has a tendency to relapse in unusual extranodal sites such as the kidney, GI tract, and ovaries, it does have a favorable prognosis when treated with combination chemotherapy often in conjunction with radiation.
FOLLICULAR LYMPHOMA, GRADE III
Grade III follicular lymphoma (>15 centroblasts/HPF) is classified as an aggressive lymphoma, since its behavior and treatment mirror that of DLBCL.
BURKITT LYMPHOMA AND BURKITT-LIKE LYMPHOMA
Small non–cleaved cell lymphomas have a characteristic cytogenetic finding, t(8;14), which occurs in about 80% of cases of Burkitt type. The c- MYC proto-oncogene on chromosome 8, thought to have a role in repressing differentiation and stimulating proliferation of cells, becomes constitutively expressed when it relocates to the immunoglobulin heavy-chain locus on chromosome 14, with reciprocal relocation of the heavy-chain gene locus to the c- MYC locus on chromosome 8. Variant translocations t(2;8) and t(8;22) involving immunoglobulin light-chain loci can be detected in the remaining 20% of cases. Classic Burkitt lymphoma is marked by small to medium-sized malignant cells having one to several distinct nucleoli, basophilic cytoplasm, and characteristic vacuoles. It is most commonly seen in pediatric patients. In Africa, patients classically present with large tumor masses of the maxilla or mandible, whereas in the United States a rapidly expanding abdominal mass is the usual presentation, occasionally accompanied by central nervous system (CNS) or bone marrow involvement.
Burkitt-like lymphoma exhibits greater cell pleomorphism than classic Burkitt lymphoma. It is composed of small to intermediate-sized immature lymphoid cells with basophilic cytoplasm and several distinct nucleoli. In patients who have undergone treatment for Hodgkin disease and in those with AIDS, this is the most common type of lymphoma occurring as a complicating factor. Unlike Burkitt lymphomas, c- MYC gene rearrangements usually do not occur in this subtype. Both Burkitt lymphoma and Burkitt-like lymphoma have a proliferation fraction (e.g., based on Ki-67 staining) close to 100%.
PRECURSOR B-LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
More common in children than adults, this disease is morphologically identical to B-cell acute lymphoblastic leukemia, and the distinction between the two is arbitrary. Lymphadenopathy is common, as is skin involvement. This disease is associated with t(9;22), which becomes progressively more common with increasing age of incidence and which is associated with a poor prognosis.
T-Cell Non-Hodgkin Lymphomas
Anaplastic Large Cell Lymphoma, T/Null Type
This distinct clinicopathologic entity, usually T-cell or null cell in phenotype, expresses CD30 (Ki-1) and, in the systemic form, is associated with a t(2;5) chromosome translocation and expression of the ALK protein. Cases of anaplastic large cell lymphoma, T/null type (ALCL) with a cutaneous presentation may be a different entity, inasmuch as they lack the t(2;5) mutation and ALK protein expression and have a clinically indolent course. It is of interest to note that the Ki-1 (CD30) antigen is also expressed on Reed-Sternberg cells in Hodgkin disease, as well as on activated B and T cells. The classic-type ALCL is characterized by a preferential perifollicular involvement of the lymph node, with sinusoidal and subcapsular infiltration. Two peaks in the age distribution have been observed—a large peak in the second to third decades and a smaller peak in the sixth to seventh decades. The disease is rapidly progressive and usually involves lymph nodes, skin, and visceral sites. ALCL may be confused with metastatic carcinoma or Hodgkin disease. The differential diagnosis, using MAbs, of several entities with similar clinical presentations is shown in Table 16.9 .
| Monoclonal Antibody | Hodgkin Disease * | Diffuse Large Cell Lymphoma | Undifferentiated Metastatic Carcinoma | |||
|---|---|---|---|---|---|---|
| T Cell | B Cell | ALCL Type † | True DHL | |||
| Ki-1 (CD30) | + | − | − | + | − | |
| CD15 (Leu-MI) | + | − | − | − | − | ± |
| T-cell markers | − | + | − | ± | − | − |
| B-cell markers | − | − | + | ± | − | − |
| LCA (CD45) | − | + | + | + | + | − |
| EMA | − | − | − | + | + | + |
* Reed-Sternberg cells (excluding the lymphocyte predominance type).
† A small percentage of T- and B-cell lymphomas are positive for Ki-1 (CD30) antigen.
PERIPHERAL T-CELL LYMPHOMA, NOT OTHERWISE SPECIFIED
This is the most common subtype of T-cell lymphoma. The tumor may express CD2, CD3, CD5, and/or CD7. CD4 is more commonly expressed than CD8. There is no characteristic cytogenetic finding, although cytogenetic abnormalities involving chromosomes 7 and 14 are common. Treatment is with combination chemotherapy as with aggressive B-cell lymphomas, although the long-term survival rate is lower.
ANGIOIMMUNOBLASTIC T-CELL LYMPHOMA
This lymphoma typically presents in older patients with a rapid onset of B symptoms (fever, night sweats, weight loss) along with a skin rash. There is a high incidence of extranodal involvement including the liver, spleen, and bone marrow. There is some heterogeneity in the disease course, however, with some patients having very indolent disease and occasionally spontaneous remissions. Concomitant immunologic problems are common, resulting in the angioimmunoblastic lymphoma with dysproteinemia syndrome with polyclonal gammopathy and such problems as autoimmune arthritis or autoimmune hemolytic anemia with a positive Coombs test. In most patients this disease is rapidly fatal, with a median survival of 12–18 months.
EXTRANODAL NK/T-CELL LYMPHOMA, NASAL TYPE
Formerly known by the less appealing “lethal midline granuloma” and also angiocentric lymphoma, this disease is most common in Asian males and is usually EBV-positive. The tumor cells often express the NK-cell marker CD56 in addition to T-cell markers. The outcome for localized disease is quite good with radiation therapy alone, but patients with disseminated disease often respond poorly to chemotherapy.
ENTEROPATHY-TYPE T-CELL LYMPHOMA
This type of T-cell lymphoma is almost always seen in the setting of celiac sprue, although the tumor cells themselves have no characteristic cytogenetic or molecular abnormality. Enteropathy-type T-cell lymphoma can present as an initial manifestation of sprue but more typically presents in patients with long-standing noncompliance with a gluten-free diet or as a sudden worsening of previously well-controlled celiac sprue. Following a gluten-free diet is extremely effective in preventing the disease but not in treating it. This is a very aggressive entity with few long-term survivors even with aggressive combination chemotherapy. Intestinal perforation and/or obstruction are common, especially in advanced disease.
SUBCUTANEOUS PANNLITIS-LIKEICU T-CELL LYMPHOMA
This is a rare lymphoma that often presents with multiple subcutaneous nodules. Since the initial course can be indolent, the disease is often misdiagnosed as benign panniculitis. Unfortunately, the disease almost invariably becomes rapidly progressive and in its later stages does not respond well to chemotherapy. For some unknown reason there is a high incidence of hemophagocytic syndrome associated with this type of T-cell lymphoma.
HEPATOSPLENIC GAMMA DELTA T-CELL LYMPHOMA
This extremely rare disease typically presents with rapid onset of hepatosplenomegaly in young males. Bone marrow involvement is common, but lymphadenopathy is generally not prominent. The neoplastic cells express the usual T-cell antigens but not the alpha beta T-cell receptor that predominates in most other forms of T-cell lymphoma. Hepatosplenic gamma delta T-cell lymphoma is extremely aggressive, and few patients survive even with aggressive chemotherapy and/or hematopoietic stem cell transplantation. A rare alpha beta variant also exists.
PRECURSOR T-LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
Common in children, particularly boys, and in young adults, precursor T-lymphoblastic lymphoma is a high-grade, immature T-cell lymphoma that is usually associated with a prominent mediastinal mass and often with SVC syndrome. It is the counterpart of childhood T-cell acute lymphoblastic leukemia, and patients in both groups are at high risk for CNS involvement. Morphologically, the malignant cells are small and uniform in appearance, showing scanty cytoplasm and indistinct nucleoli. Characteristic nuclear convolutions are usually present; mitoses are frequent.
ADULT T-CELL LYMPHOMA-LEUKEMIA
This unusual T-cell malignancy is composed of neoplastic lymphoid cells of various sizes, having irregular nuclei, some containing nucleoli, often with marked convolutions of the nuclear contours. Lymph node biopsy characteristically shows a leukemic pattern of infiltration. Seen predominantly in Japan and in blacks of the West Indies, the Caribbean nations, and the southeastern United States, adult T-cell lymphoma-leukemia (ATLL) typically shows rapid progression, with early involvement of lymph nodes, skin, bone, blood, and bone marrow. Hypercalcemia often develops. The lung, liver, GI tract, and CNS may also be involved. The disease is caused by a C-type RNA retrovirus known as HTLV-1, infection with which often occurs many decades before the development of ATLL.
MISCELLANEOUS LYMPHOMAS
Extranodal Lymphomas
NHLs often secondarily invade visceral sites, resulting in stage IV disease. Primary involvement of extranodal sites is less common, occurring in up to 25% of cases. Extranodal lymphomas may arise in almost any site, particularly the skin, thyroid, orbit, Waldeyer’s ring, CNS, lung, GI tract, reproductive organs, breast, stomach, and kidneys. The vast majority of these cases respond to combination chemotherapy, with or without local radiation therapy.
Lymphoma of the Breast
Primary lymphomas of the breast, which are rare, may present as an expanding breast mass, clinically simulating breast carcinoma. Therefore, biopsy specimens must be carefully examined, with the use of surface marker studies when possible, to avoid an unnecessary mastectomy. Most lymphomas of the breast are derived from B cells and are DLCL or marginal zone lymphoma, although rare cases of follicular lymphoma and other types have been reported. For reasons that remain unclear, patients with primary DLBCL of the breast are at increased risk for involvement of the CNS and must receive intrathecal chemotherapy or other chemotherapeutic prophylaxis to prevent the development of CNS disease.
LYMPHOMA OF THE TESTICLE
Most lymphomas of the testicle are high-grade lesions—DLBCL, lymphoblastic lymphoma, or Burkitt lymphoma. Involvement of the testicle with DLBCL is a risk factor for involvement of the CNS, and these patients must receive intrathecal chemotherapy or other chemotherapeutic prophylaxis to prevent the development of CNS disease. Also, because the blood-testis barrier can prevent chemotherapy from effectively penetrating into testicular tissue, these patients often receive adjuvant irradiation to the contralateral testicle.
LYMPHOMA OF THE GASTROINTESTINAL TRACT
The GI tract is the most common site of primary extranodal lymphoma, which accounts for approximately 2% of GI neoplasms and most commonly involves the stomach followed by the small intestine and then the colon. Approximately 5% of gastric neoplasms are malignant lymphomas, the majority of which are DLBCL. They most commonly occur in middle or late adulthood, typically arising in the body of the stomach; they are often large and diffuse or may appear as nodular masses. Direct spread to the liver or spleen, as well as dissemination to other areas, occasionally occurs. Gastric lymphomas carry a better prognosis than gastric carcinomas; the 5-year survival rate is approximately 50% for all stages.
Extranodal marginal zone lymphomas of MALT account for a significant percentage of GI lymphomas and also occur in several other extranodal sites that normally contain MALT, including breast, lung, head, and neck areas, including the thyroid, and the urogenital tract. It may be low grade or high grade. Low-grade MALT lymphoma is composed of small B cells with round to irregular nuclear outlines, notable cytoplasm, associated plasma cells, and a tendency to invade epithelium, resulting in characteristic lymphoepithelial lesions. Tumor cells are immunoreactive for pan-B-cell markers (e.g., CD20) but not CD5, seen in other low-grade B-cell lymphomas, or CD10, seen in follicular center cell lymphomas. Low-grade MALT lymphomas tend not to disseminate widely, and surgical excision results in an excellent long-term prognosis. Of note, NHL (particularly low-grade MALT type) affecting the stomach but not other sites is associated with previous H. pylori infection ( ). Despite lymphoma regression with therapy for H. pylori , the causative role, while plausible, still remains unproven ( ).
LYMPHOMA OF THE KIDNEY
Although primary renal lymphoma is rare, about 5% to 10% of patients with disseminated lymphoma show clinically detectable renal involvement and up to 50% of cases reveal renal lesions at autopsy. High-grade lymphomas are most commonly encountered, particularly Burkitt lymphoma. Occasionally patients present with renal failure due to infiltration of the kidney by lymphoma cells, with resultant bilateral enlargement.
INTRAVASCULAR LYMPHOMA
Historically also referred to as malignant angio-endotheliomatosis, this rare B- or T-cell–derived neoplastic proliferation of large pleomorphic mononuclear cells may preferentially involve the lumens of small arteries and veins, and capillaries, without significant infiltration of vessel walls or parenchymal involvement that may be seen in angioimmunoblastic T-cell lymphoma. Intravascular lymphoma typically involves multiple organs, with effects particularly notable in the CNS (with associated dementia and neurologic impairment) and skin (with plaques and nodules), and is usually rapidly fatal.
CLINICAL EVALUATION AND STAGING
The staging workup for all patients includes a detailed history and physical examination; complete blood count with differential; blood chemistries; serum protein electrophoresis; chest radiograph (now largely replaced by CT scanning); CT scan of the chest, abdomen, and pelvis; and bone marrow aspiration with biopsy. Selected patients may require the following additional studies for complete evaluation: magnetic resonance imaging (MRI; to evaluate the nervous system or bone), GI series or endoscopy, bone scan, CT scan of the head (in the presence of symptoms of lymphoma in the head and neck area), and lumbar puncture. Evaluation of the peripheral blood by a sensitive cytofluorometric technique (cell sorter) may reveal circulating monoclonal lymphocytes, especially in B-cell lymphomas. As with Hodgkin disease, PET scans have largely replaced gallium scans and have become relatively routine for the staging and therapeutic monitoring of NHL (particularly the aggressive subtypes).
The Ann Arbor staging system is used for NHLs, except that the designation of stages III 1 and III 2 as applied to Hodgkin disease is not necessary (see Fig. 16.13 ). This system, however, is not optimal, for it does not reflect important prognostic factors such as size, or “bulk,” of the tumor, the number of extranodal sites of disease, performance status, or the lactate dehydrogenase (LDH) concentration. Separate staging classification systems have been devised for childhood lymphomas, particularly Burkitt lymphoma and lymphoblastic lymphoma.
CLINICAL MANIFESTATIONS
In contrast to Hodgkin disease, most patients with NHL present with advanced disease (stage III or IV), which is often reflected by generalized adenopathy. In addition, the disease may occur at unusual sites, such as epitrochlear or popliteal nodes, Waldeyer’s ring (nasopharynx), skin, GI tract, brain, and ovaries or testes—sites rarely affected in Hodgkin disease. Patients with low-grade lymphomas have a long history of slowly progressing disease, which may temporarily regress—so-called “spontaneous” remission—in 5% to 10% of cases. High-grade and aggressive intermediate-grade lymphomas, on the other hand, usually have a more rapidly progressive course, with 40% to 50% of patients developing B symptoms. Although the presence of B symptoms has been considered an adverse prognostic factor, multivariate analysis after modern intensive chemotherapy programs reveals that B symptoms no longer affect survival.
Bone marrow involvement occurs in 10% to 40% of cases. The incidence is highest in lymphoblastic, follicular, and Burkitt and Burkitt-like lymphomas (20% to 40%) and lowest in DLBCL (10% to 15%). Such involvement is often focal and does not affect peripheral blood counts, although in some patients the marrow may be extensively infiltrated, leading to the development of pancytopenia or a leukemic phase. Immunofluorescent microscopy has revealed small numbers of monoclonal B cells in the peripheral blood in about one third of patients with low-grade lymphomas and in 15% to 20% of those with intermediate- and high-grade lymphomas.
PROGNOSTIC FACTORS
Modern combination chemotherapy programs have markedly improved the prognosis, particularly in intermediate- and high-grade lymphomas. While formerly only 10% of patients were cured, 50% to 60% are now cured with intensive multidrug regimens. As a result of the clinical, histologic, and immunobiologic heterogeneity of NHL, many prognostic factors have been identified that separate patients into various risk categories for relapse and decreased survival. This allows for planning of lesser or greater intensity of treatment (see Table 16.2 ).
| Host/Tumor Characteristics | Clinical Prognostic Factors |
|---|---|
| Tumor’s growth and invasive potential | Serum LDHNumber of nodal and extranodal sites of diseaseMass sizeStage according to Ann Arbor classificationBM involvement |
| Patient’s response to the tumor | Systemic B symptoms |
| Performance status | |
| Patient’s ability to tolerate intensive therapy | Age at diagnosis |
| Performance status | |
| BM involvement |
* Aggressive lymphomas refer to NCI categories E to H; see Table 16.1 .
A prognostic index has been developed by the International Non-Hodgkin Lymphoma Prognostic Factors Project based upon patient data from 16 single institutions and cooperative groups ( ). A total of 2031 evaluable patients with aggressive NHL (NCI categories E to H; see Fig. 16.35 ) were treated with doxorubicin-containing regimens between 1982 and 1987. Multiple clinical and laboratory features were analyzed to define adverse prognostic factors that were present at the time of diagnosis and predicted for relapse and death. The International Index Model, based upon age, stage, serum LDH, performance status, and number of extranodal disease sites, identified four risk groups with predicted 5-year survivals of 73%, 51%, 43%, and 26% (see Tables 16.3 and 16.4 ). It was found that while older patients (>60 years) had similar complete remissions (CRs) to younger patients (<60 years), they were less likely to maintain their CR than younger patients, especially low- and low- to intermediate-risk patients, with resultant shorter survival. Survival curves for the age-adjusted International Index are noted in Figure 16.36 . The advantage of a prognostic index is that good-risk patients can be identified for conventional therapy while poor-risk (high-relapse) patients can be identified for new research protocols to improve the cure rate. Furthermore, treatment results among institutions and cooperative groups can be compared, since standardized prognostic factors will have been used. FLOAT NOT FOUND
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


