Bone sarcomas can occur through the musculoskeletal system. Osteosarcomas are more prevalent in the appendicular skeleton, with the most prevalent sites being in the distal femur (32%), proximal tibia (15%), proximal humerus (8%), and proximal femur (5%).39 This distribution corresponds to the relative activity of the physeal plates in the growing skeleton. By contrast, Ewing sarcoma favors the axial skeleton, with the pelvis/sacrum (22%), proximal femur (10%), shoulder girdle (12%), and ribs (8%) being the most common locations, followed by the long bones and sacrum.39 Chondrosarcoma similarly occurs most frequently in central regions: pelvis/scrum (26%), shoulder girdle (14%), proximal femur (12%), and ribs (11%).39 When found in the long bones, the epicenter of conventional osteosarcoma lesions tends to be in the metaphysis, with diaphyseal extension, whereas Ewing sarcoma often arises in the diaphysis with metaphyseal extension. Juxtacortical osteosarcoma and chondrosarcoma variants occur on the bone surface.
Some primary bone malignancies manifest in characteristic anatomic locations. Clear cell chondrosarcoma, for instance, most often arises in the femoral head.40 Parosteal osteosarcoma favors the posterior distal femur.41 Periosteal osteosarcoma is found in the anterior tibial shaft.42 Adamantinoma is most frequently encountered in the tibia and/or fibula.43 Chordoma has a predilection for the sacrum and the clivus.1,44
Finally, several histologic equivalents of bone sarcomas can be found in soft tissues and joints. Extraskeletal osteosarcoma is a rare neoplasm that has a median survival of 46 months for patients with localized disease.45 By contrast, extraosseous Ewing sarcoma has a prognosis similar to its primary bone counterpart.46 Extraskeletal myxoid chondrosarcoma, also an aggressive soft tissue lesion with high rates of local/distant recurrence and delayed disease-associated death, is a misnomer insofar as the tumor is really of uncertain differentiation with no convincing evidence of cartilaginous differentiation.47,48 As a rule, it is exceedingly rare for a sarcoma of bone or soft tissue to occur in an intra-articular location, but chondrosarcomas of the synovium, arising de novo or secondary to synovial chondromatosis, have been described.49
Gross anatomic pathology varies widely according to histologic subtype, grade, and anatomic location. Although the soft tissue component of many high-grade sarcomas of bone have a characteristic whitish, firm, “fish flesh” appearance, gross findings can run the gamut from heavily ossified areas to myxoid, friable, or frankly necrotic regions, even within the same specimen. As most high-grade bone sarcomas are >5 cm and associated with a soft tissue mass that is extracompartmental to bone, the tumor classification according to the Enneking system50,51 is most often “T2b.”
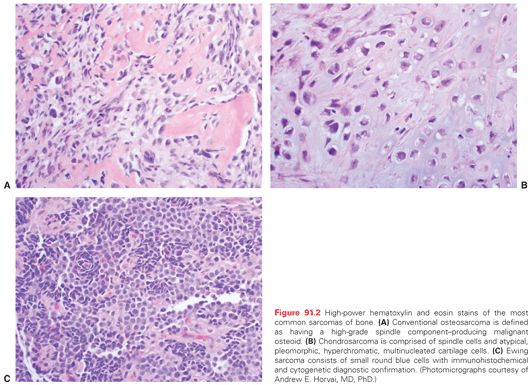
Microscopic pathology also varies considerably amongst the bone sarcomas. The prototypical bone sarcoma, a conventional high-grade osteosarcoma, consists, in most basic terms, of a spindle cell neoplasm that produces osteoid (Fig. 91.2A). A high-grade chondrosarcoma will have a spindle cell component admixed with malignant cartilaginous matrix (Fig. 91.2B). By contrast, Ewing sarcoma is a small round blue cell neoplasm for which the definitive diagnosis is suggested by immunohistochemical staining with CD-99 and confirmed by cytogenetic results of t(11:22) chromosomal translocation (Fig. 91.2C).
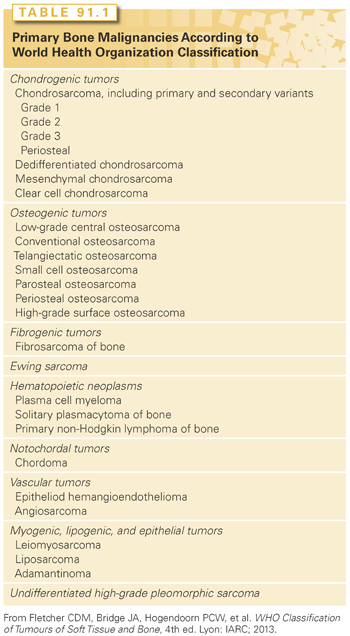
From a pathologic standpoint, the World Health Organization recognizes >25 different types of primary bone malignancies.48 With the exception of plasma cell myeloma, primary non-Hodgkin lymphoma of bone, chondrosarcoma, conventional osteosarcoma, and Ewing sarcoma, the remaining entities are, for the most part, very rare (Table 91.1).
There exist no population-based screening tests applicable to the detection of bone sarcomas. However, a number of syndromic conditions that predispose to the development of primary bone malignancies are known (Table 91.2). Many of the implicated genetic diseases, such as Rothmund-Thomson and Werner syndromes, are exceedingly rare, and no clear algorithms exist for cancer screening. The same is true for more commonly encountered conditions that predispose to bone cancer, such as Li-Fraumeni syndrome, for which a recent workshop concluded, “further tests are needed to test screening protocols . . . which would be aimed at establishing effective screening regimens that take into account the wide diversity of disease phenotypes in TP53 mutation carriers.”11 For patients with predisposition diagnoses that are relatively commonly encountered in orthopedic oncology offices, such as hereditary multiple hereditary exostoses, multiple enchondromatoses, polyostotic fibrous dysplasia, and Paget disease, several practical, generalized guidelines can be offered. Plain radiographs of bones that are moderately to severely affected should be obtained yearly. A limited skeletal survey of involved areas (including, as indicated, anteroposterior [AP] and lateral views of the humeri and femora; AP views of the pelvis and distal extremities) should be considered every 2 years. Magnetic resonance (MR), computed axial tomography (CT), and positron emission tomography (PET) screening should be considered based upon a patient’s family and personal clinical history. In all circumstances, care should be taken to limit radiation exposure with screening tests. However, as an individual ages, sarcoma risk increases, and so routine surveillance surveys should be offered, especially for deep-seated locations such as the pelvis and shoulder girdle, where most sarcomas in these conditions will become manifest.33 Because malignancies in these deep-seated areas can become relatively large before symptoms develop, vigilance of occult locations is especially warranted.

It is nearly universally true of practice patterns within the United States that patients suspected of being at risk for having a bone sarcoma have first had a plain radiographic examination ordered by their initial point of contact within the health-care system. It is also very common for the primary care provider or general orthopedic surgeon to obtain an MR scan of the affected area (Table 91.3). But once concern for the possibility of a bone sarcoma has been raised, further diagnostic tests, many of which are of a specialized nature and involve radiation exposure, should be deferred, and the patient should be immediately referred to a multidisciplinary center skilled in sarcoma management.1

History
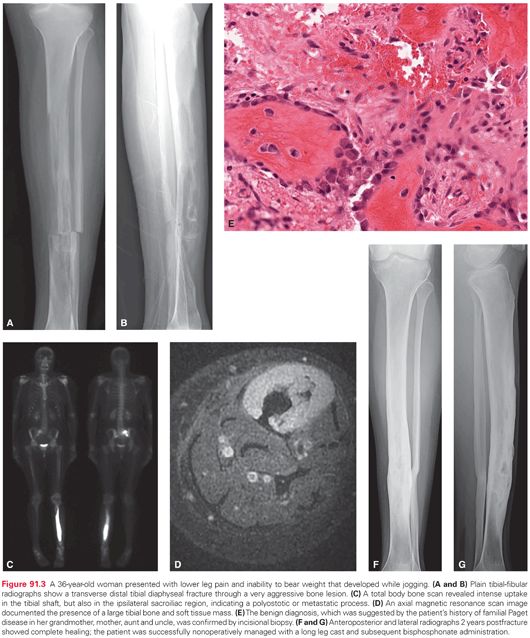
Good medicine starts with a thorough history. Aggressive bone lesions often give rise to pain before a mass is noted. The nature of the discomfort, in terms of its intensity, frequency, duration, pattern, localization, and aggravating/alleviating factors must be carefully assessed. The patient should be questioned regarding associated symptoms such as numbness, tingling, weakness, stiffness, instability, and gait abnormalities. Documentation of the characteristics of a mass, if present, must be obtained with respect to time course, growth pattern, and presence of localized erythema, warmth, and tenderness. Systemic findings such as fever, sweats, chills, weight loss, and fatigue need to be queried. A past personal and/or family medical history of conditions that predispose to bone malignancies must be explored. In many cases of even high-grade sarcomas, the history is that of an enlarging bone and soft tissue mass that is less painful than might be expected. Knowledge gained regarding history can direct further evaluation away from or toward nonneoplastic, pseudotumorous, aggressive, metabolic, or secondary conditions that can mimic primary bone malignancies, such as osteomyelitis, eosinophilic granuloma, giant cell tumor of bone, osteoblastoma, metastatic disease, or Paget disease (Fig. 91.3).
Physical Examination
Although it may not narrow the differential list considerably, a thorough physical examination remains an essential part of the diagnostic pathway for sarcomas of bone. Café-au-lait spots can signal the presence of fibrous dysplasia; the integrity of the integumentary system should be assessed for erythema, warmth, and drainage over the primary site. The size, location, mobility, and consistency of any mass, if present, should be carefully described. The presence of absence of regional lymphadenopathy must be noted. Orthopedic parameters, including limb length, active and passive ranges of motion, joint stability, and gait pattern, need to be reviewed. Finally, a pertinent neurovascular exam must be completed.
Diagnostic Studies
Though neither highly sensitive nor specific, laboratory testing should not be neglected in the evaluation of patients suspected of having a sarcoma of bone. A complete blood count, erythrocyte sedimentation rate, and C-reactive protein level can be helpful in ruling out infection. Alkaline phosphatase and lactic dehydrogenase are sometimes elevated in skeletal sarcomas; creatinine and calcium levels are also useful chemistry tests. Serum and urine immunoelectrophoretic analyses can assist in excluding myeloma, and a basic urinalysis can be used to screen for genitourinary health.
Radiologic imaging represents the bulk of diagnostic studies for aggressive bone conditions. Plain roentgenograms include orthogonal views of the primary skeletal site and two views of chest as an initial assessment of metastatic pulmonary disease. Nuclear medicine tests include technetium-99m total body bone, indium-labeled white blood cell, and PET scans. A bone scan aids in determination of primary site activity, and the presence or absence of polyostotic or metastatic disease. An indium scan can examine the possibility of osteomyelitis. A PET or PET/CT scan quantifies metabolic activity in the primary site and helps to exclude occult metastases. The utility of [F-18]-fluorodeoxy-D-glucose PET/CT scanning in the management of patients with bone sarcoma is well established, and should be considered to be a care standard.52–58 A dedicated chest CT scan is perhaps the most sensitive way to exclude distant disease in the lungs, which is the most common site of metastatic deposits in patients with sarcoma. CT scanning also has proven utility in studying axial primary sites and juxtacortical lesions, as well as in predicting pathologic fracture risk.59 Often the best modality for documenting the extent of a primary bone and soft tissue mass is an MR scan, which should include the entire involved bone so as to make sure that skip metastases are not present. Specialized cross-sectional studies, including MR neurograms, MR angiograms, and CT angiograms are sometimes important to assist with surgical planning.
In the case of suspected primary bone malignancy, supreme caution must be taken with respect to the manner in which the biopsy is undertaken. Percutaneous fine needle or core procedures, especially when guided by imaging such as ultrasound, CT, or MR scanning, can be successful in establishing a diagnosis even in deep anatomic locations, especially if a soft tissue mass is present; this method has the advantage maximizing sampling throughout the mass while minimizing contamination.60–67 As sufficient material must be obtained to perform all histologic, immunohistochemical, cytometric, and cytogenetic studies, thereby allowing an accurate diagnosis, open (incisional) biopsies are often warranted, especially in pediatric cases where multi-institutional protocol studies request centralized pathologic review and other specialized testing. Excisional (resection) biopsy can be considered for smaller lesions that can be completely excised with negative margins and without undue functional compromise; an atypical or low-grade chondrosarcoma arising in an osteochondroma is an example of this type of biopsy procedure. Whenever an open biopsy procedure is chosen, careful attention must be paid to incisional length (short) and placement (in line with the definitive resection procedure), dissection planes (through rather than between muscular planes), and avoidance of neurovascular exposure, bleeding, and infection.68–71 A skilled musculoskeletal pathologist should be immediately available to review the frozen section, confirm the receipt of sufficient biopsy material, and perform direct handling of the specimen.71 Improperly performed biopsies can delay the start of care, prompt inappropriate treatment, preclude limb salvage by causing infection or contamination, and otherwise result in local recurrence and amputation. The risk of diagnostic errors and complications increases by as much as 12-fold when the biopsy is improperly done.69 Because of these grave medical and medicolegal concerns, referral to a tertiary center skilled in the multidisciplinary bone sarcoma management prior to biopsy is strongly advised.1
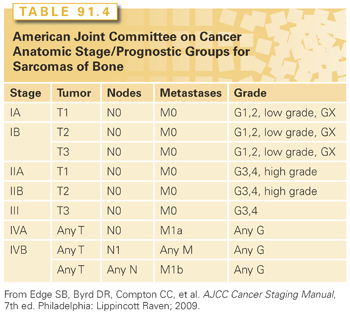
Once all diagnostic studies are complete, formal oncologic staging concludes the pretreatment evaluation process. Staging should be performed according to TNM (tumor, node, metastasis) guidelines with principles set forth by the American Joint Committee on Cancer.72 For bone sarcomas, staging correlates directly with prognosis, which is of utmost importance when discussing expectations regarding outcomes with patients and families (Table 91.4).
MANAGEMENT BY DIAGNOSIS AND STAGE
Systemic Therapies for Ewing Sarcoma
Chemotherapy for Patients with Newly Diagnosed Ewing Sarcoma
Prior to the routine use of chemotherapy, nearly all patients with newly diagnosed Ewing sarcoma developed distant metastatic disease and ultimately died.73–75 These observations underscored the nearly universal presence of occult micrometastatic disease in patients with Ewing sarcoma. While local control measures (surgery and/or radiotherapy) are critical to the treatment of patients with Ewing sarcoma, systemic therapy is equally critical to attaining cure.
Early single-institution studies suggested that the regimen of vincristine, doxorubicin, actinomycin-D, and cyclophosphamide (VACA) was active in Ewing sarcoma.74–76 Follow-up cooperative group trials established the efficacy of VACA and began to refine this regimen for Ewing sarcoma. For example, the first North American Intergroup Ewing Sarcoma Study (IESS-I) investigated the role of doxorubicin and whole lung radiation in the context of vincristine, dactinomycin, and cyclophosphamide.77 Patients with newly diagnosed disease were randomized to one of three arms: vincristine, dactinomycin, and cyclophosphamide (VAC); VAC plus doxorubicin (VACA); or VAC with whole lung radiation. Among 342 eligible patients, patients randomized to VACA had significantly superior relapse-free survival compared to patients randomized to the other two arms. Patients randomized to receive VAC had the worst outcomes, and patients randomized to receive VAC with whole lung radiation had intermediate outcomes. These results definitively established the importance of doxorubicin in the management of Ewing sarcoma, but also demonstrated that whole lung radiation may prevent some cases of relapse even in the absence of documented lung metastasis.
Other national groups adopted the VACA regimen, with largely similar results. For example, the CESS-81 trial was a nonrandomized trial evaluating VACA in patients with localized Ewing sarcoma.78 This group reported a 5-year disease-free survival rate of 54%. In addition, they identified large tumor size and poor histologic necrosis to neoadjuvant chemotherapy as potential adverse prognostic factors in this setting. The UK Children’s Cancer Study Group conducted a similar trial, though the doxorubicin dose intensity was lower than in other trials.79 They reported a 5-year relapse-free survival rate of 41% and suggested that this inferior outcome may be due to lower doxorubicin dose intensity. The French Society of Pediatric Oncology also treated patients with VACA and reported a 4-year disease-free survival rate of 52%.80
Subsequent trials have built upon the success obtained with VACA. Strategies have included changing the dosing strategy in VACA and adding ifosfamide or ifosfamide/etoposide (IE). The French EW88 trial used this first approach to study a more protracted VACA treatment schedule in which cyclophosphamide was delivered daily for 7 days each cycle, with cycles repeated at 2-week intervals.81 In this nonrandomized trial, outcomes appeared to be superior compared to previous trials, though more intensive local control measures were also prescribed in this trial. Nevertheless, these results suggested that more frequent chemotherapy cycles were beneficial and/or that more protracted cyclophosphamide exposure was beneficial.
The second Intergroup Study (IESS-II) also sought to clarify the optimal mode of administration of VACA.82 Patients with newly diagnosed localized nonpelvic disease were randomized to one of two different VACA schedules. In the intensive schedule, patients received higher doses of doxorubicin and cyclophosphamide given in cycles administered every 3 weeks. In the protracted schedule, patients received lower doses of these agents and exposure of the cyclophosphamide was distributed across 6 sequential weeks. This design resulted in significantly greater early doxorubicin exposure in the intensive arm as well as a slightly higher cumulative doxorubicin exposure in that arm. Among 214 eligible patients, all outcome measures (relapse-free, disease-free, and overall survival) were superior for patients randomized to the intensive arm. These results emphasized the importance of early doxorubicin intensity and led to adoption of VACA administration in 3-week cycles as a standard approach in subsequent studies.
Based upon response rates of approximately 30% in patients with recurrent Ewing sarcoma, several groups investigated the role of ifosfamide or IE in patients with newly diagnosed disease. The French cooperative group conducted a nonrandomized trial in which ifosfamide replaced cyclophosphamide throughout neoadjuvant and adjuvant chemotherapy that also contained vincristine, doxorubicin, and dactinomycin.83 Outcomes of the 65 localized patients treated with this strategy were compared to 95 patients treated on a previous French trial in which all patients received cyclophosphamide. Replacing cyclophosphamide with ifosfamide did not appear to improve outcomes compared to historic controls, suggesting that ifosfamide (without etoposide) is not active in this setting or that eliminating cyclophosphamide exposure altogether was deleterious.
A nonrandomized trial from the UK Children’s Cancer Study Group used a similar approach to study ifosfamide, but yielded a different result.84 A total of 201 patients with localized disease were nonrandomly assigned to receive therapy with ifosfamide replacing cyclophosphamide. Compared to historic controls treated with cyclophosphamide, relapse-free and overall survival appeared more favorable with the use of ifosfamide.
The CESS-86 trial used a similar approach, though in a risk-stratified manner.85 Patients with small, localized extremity tumors received VACA, whereas patients with either large tumors or axial tumors had ifosfamide substituted for cyclosphosphamide (VAIA). Although patients treated with VAIA were predicted to have inferior outcomes due to adverse baseline risk factors, event-free survival was similar compared to patients with lower risk disease treated with VACA. These results suggested that the use of ifosfamide helped to mitigate the adverse impact of large tumor size and/or axial tumor site.
An Italian Cooperative Group trial nonrandomly evaluated the addition of ifosfamide to VACA in the neoadjuvant setting and the addition of IE in the adjuvant setting.86 The trial included 160 patients with localized disease. Event-free and overall survival estimates were among the highest reported at that time, suggesting that ifosfamide and/or IE may have improved outcomes in this population.
The most definitive trial on the role of IE was North American Intergroup trial INT-0091.87 Patients with newly diagnosed Ewing sarcoma were randomized at study entry to receive either standard therapy with vincristine, doxorubicin, and cyclophosphamide (VDC) administered every 3 weeks or VDC cycles alternating every 3 weeks with IE cycles. All patients received the same duration of therapy and the same cumulative dose of doxorubicin, with actinomycin-D substituted once patients received that cumulative doxorubicin dose. A total of 518 eligible patients were randomized, 398 of whom had localized disease at study entry. Among patients with localized disease, there was a statistically significant difference in 5-year event-free survival (54% for patients randomized to VDC versus 69% for patients randomized to VDC/IE). The addition of IE to VDC did not improve outcomes for patients with metastatic disease at initial presentation.87,88 This result established VDC/IE as a new North American standard of care for patients with newly diagnosed localized Ewing sarcoma.
The next generation of clinical trials from the Children’s Oncology Group (COG) has sought to intensify the VDC/IE regimen. Intergroup trial INT-0154 was a randomized trial for patients with newly diagnosed localized Ewing sarcoma.89 Patients were randomized at study entry to receive standard dose VDC/IE every 3 weeks or higher-dose VDC/IE. Patients in the experimental arm received higher doses of ifosfamide and cyclophosphamide during cycles of chemotherapy. All patients received approximately the same cumulative doses of chemotherapy such that patients on the experimental arm completed therapy earlier than patients on the standard arm. A total of 478 eligible patients were randomized. There was no statistically significant difference in event-free survival between the standard arm and the experimental arm, demonstrating that higher-dose therapy did not improve outcomes.
COG protocol AEWS0031 was also a randomized trial for patients with newly diagnosed localized Ewing sarcoma.90 This trial sought to intensify therapy not by dose escalation, but rather by decreasing the interval between chemotherapy cycles (interval compression). Patients were randomized at study entry to receive standard therapy with VDC/IE cycles alternating every 3 weeks or to the experimental arm with VDC/IE cycles alternating every 2 weeks. A total of 568 eligible patients were randomized. Patients randomized to the interval-compressed arm had a significantly greater 5-year event-free survival (73% versus 65% for patients randomized to the standard arm). This trial established interval compressed VDC/IE as a new standard approach for patients with localized Ewing sarcoma. Of note, the role of interval compression in patients with newly diagnosed metastatic Ewing sarcoma has not been evaluated.
The European Intergroup Cooperative Ewing’s Sarcoma Study (EICESS)-92 trial sought to clarify the role of ifosfamide and etoposide based upon a risk classification strategy in which patients with small, localized tumors were deemed standard risk and patients with large localized tumors or any metastases were deemed high risk.91 All standard-risk patients received initial therapy with VAIA. These patients were then randomized to receive this same therapy in the adjuvant setting or this same therapy with cyclophosphamide substituting for ifosfamide. Among 155 randomized standard-risk patients, there was no difference in event-free or overall survival between patients who received ifosfamide or cyclophosphamide in the adjuvant setting. These results do not fully clarify the role of ifosfamide in this group as all patients received ifosfamide in the neoadjuvant setting.
High-risk patients in the EICESS-92 trial were randomized at enrollment to receive VAIA or VAIA plus etoposide.91 Among 492 randomized high-risk patients, there was a trend favoring the addition of etoposide. In particular, patients deemed high-risk due to large localized tumors appeared to benefit from the addition of etoposide, whereas patients with metastatic disease did not appear to benefit from etoposide. These results mirror those obtained from INT-0091 in which the addition of IE benefited patients with localized disease but not patients with metastatic disease.
Based upon this current body of evidence, a standard approach for newly diagnosed patients with localized Ewing sarcoma in North America is to use interval compressed VDC/IE according to AEWS0031. Many clinicians utilize this same regimen for patients with newly diagnosed metastatic disease, though data supporting a benefit of IE or of interval compression are lacking in this population. For both groups, participation in a cooperative group clinical trial should be strongly considered.
Chemotherapy for Patients with Recurrent Ewing Sarcoma
Historically, patients with recurrent Ewing sarcoma had few systemic options. Many such patients were retreated with chemotherapy combinations used as part of initial therapy. One series reported re-responses and durable remissions in patients treated with this strategy at relapse.92 Additional data suggest that higher dose ifosfamide (15 g/m2) may be active in patients with recurrent Ewing sarcoma who were treated with lower doses of ifosfamide as part of initial therapy.93 Given greater options, many clinicians now opt to treat patients with new agents not previously used as part of initial therapy.
Currently, patients with recurrent Ewing sarcoma are candidates for clinical trials of novel agents or may be treated with a number of salvage chemotherapy regimens with documented activity in this setting. The combination of gemcitabine with docetaxel has shown modest activity in patients with recurrent Ewing sarcoma. Two single-institution case series reported no responses among four patients with recurrent Ewing sarcoma.94,95 A formal phase 2 trial of standard doses of gemcitabine and docetaxel reported 2 patients with partial responses among 14 patients with recurrent Ewing sarcoma.96 In contrast, another case series reported four of six patients with recurrent Ewing sarcoma with objective responses with the use of higher-dose gemcitabine and docetaxel, suggesting that higher doses of this regimen may have greater activity in this disease.97 A formal phase 2 trial of higher-dose gemcitabine together with standard-dose docetaxel reported one patient with a partial response among seven patients with recurrent Ewing sarcoma.98 Taken together, these findings suggest a limited role for this regimen in the management of patients with recurrent Ewing sarcoma.
Campothecin-based regimens are currently the most active available chemotherapy regimens for patients with relapsed Ewing sarcoma. The combination of topotecan with cyclophosphamide has shown activity in this population. A Pediatric Oncology Group (POG) phase 2 trial of this combination included 17 patients with relapsed Ewing sarcoma, 2 with complete response, and 4 with a partial response for an objective response rate of 36%.99 A retrospective analysis from the German cooperative group included 49 patients with relapsed Ewing sarcoma and disease evaluable for response.100 On initial response assessment, 16 (33%) had a partial response. Based upon these findings in patients with recurrent Ewing sarcoma, the COG conducted a trial in patients with newly diagnosed metastatic Ewing sarcoma in which patients received an initial window of therapy with either two courses of topotecan monotherapy or topotecan plus cyclophosphamide.101 Topotecan monotherapy had only modest activity, but two courses of the combination of topotecan plus cyclophosphamide resulted in a response rate of 57%. Based upon these promising results in patients with relapsed or metastatic Ewing sarcoma, the current COG trial for patients with newly diagnosed localized disease (AEWS1031) is a randomized comparison of interval compressed VDC/IE to interval compressed VDC/IE with the addition alternating blocks of topotecan and cyclophosphamide (NCT01231906).
The combination of irinotecan and temozolimide has also shown activity in patients with relapsed Ewing sarcoma. In the initial pediatric phase 1 trial of this regimen, three of seven patients with Ewing sarcoma had objective responses.102 A multicenter retrospective analysis of 14 patients treated with this regimen showed a response rate of 29%.103 Follow-up retrospective studies from other institutions reported response rates >60% in this same population.104,105 This combination is less myelosuppressive than topotecan/cyclophosphamide and may be given as an oral regimen. Given these properties, this combination may serve as a backbone for the development of new regimens that combine chemotherapy with novel agents.
Role of High-Dose Chemotherapy for Patients with Ewing Sarcoma
Given the finding that Ewing sarcoma is a chemosensitive tumor, several groups have evaluated the role of high-dose chemotherapy with autologous stem cell rescue in selected patients with high-risk disease. A single-institution retrospective study reported on the role of this approach in patients with recurrent Ewing sarcoma.106 On univariate analysis, patients who received high-dose chemotherapy had superior overall survival. In order to try to control for selection bias that may have led to selection of the most favorable patients to receive high-dose chemotherapy, this group performed a multivariate survival analysis. Receipt of high-dose chemotherapy as a component of relapse therapy was associated with improved survival, even after controlling for variables thought to be prognostic at relapse, such as response to salvage therapy and time from diagnosis to relapse.
The majority of studies evaluating this modality have focused on patients with newly diagnosed metastatic Ewing sarcoma. Given the radiosensitivity of Ewing sarcoma, the Children’s Cancer Group conducted a trial evaluating high-dose therapy using melphalan, etoposide, and total-body radiation as conditioning.107 Thirty-two patients with newly diagnosed disease with metastases to the bone and/or bone marrow were nonrandomly assigned to high-dose therapy following induction chemotherapy. Event-free survival was 20% at 2 years and was identical to a historic control group treated with standard-dose chemotherapy, indicating that this regimen did not improve outcomes in this very high-risk patient population.
A series of multicenter trials treated patients with high-dose therapy with busulfan and melphalan conditioning.108,109 This approach appeared to be feasible following initial induction chemotherapy, though whether outcomes were improved by this approach is not clear in these uncontrolled trials. The Euro-Ewing group applied this approach to a larger group of patients with metastatic disease beyond isolated lung metastases.110 In this very high-risk patient population, 3-year event-free survival was 27%, suggesting that any improvement in outcome from high-dose therapy was incremental.
One trial has been completed evaluating the role of high-dose chemotherapy in patients with high-risk localized Ewing sarcoma.111 Patients received a common induction chemotherapy regimen and had response assessed at the time of local control (by histology for patients who underwent surgical local control or by imaging for patients who received radiation for local control). Patients with a poor response to induction chemotherapy were nonrandomly assigned to receive high-dose chemotherapy with busulfan and melphalan conditioning. Only a subset of patients assigned to receive high-dose chemotherapy did so. This selected subset had an event-free survival rate that was similar to patients with a good response who received ongoing standard-dose chemotherapy. Patients with a poor response to induction who did not go on to receive high-dose therapy had significantly inferior outcomes. These results suggest that high-dose therapy may have abrogated the anticipated poor outcomes for patients with a poor response to induction chemotherapy.
A lingering concern and area of controversy in this field is that patients with chemotherapy-responsive disease are both more likely to survive longer and more likely to be candidates for high-dose chemotherapy. In order to circumvent this selection bias, the Euro-Ewing group is conducting the first randomized trial of high-dose chemotherapy in this disease. Patients with newly diagnosed Ewing sarcoma and either isolated lung metastases or localized disease with poor response to initial chemotherapy are eligible. All patients receive induction chemotherapy with vincristine, ifosfamide, doxorubicin, and etoposide.112 Patients randomized to undergo high-dose chemotherapy receive conditioning with busulfan and melphalan. Patients randomized to receive ongoing chemotherapy receive the combination of vincristine, doxorubicin, and ifosfamide. Until the results of this ongoing randomized trial become available, the role of high-dose chemotherapy for patients with Ewing sarcoma will remain controversial.
Novel Agents for Patients with Ewing Sarcoma
Despite understanding the critical role of EWSR1 fusion oncogenes in the pathogenesis of Ewing sarcoma, strategies to target EWSR1 fusion oncogenes and oncoproteins have been difficult to develop. One group used gene expression profiling to identify existing drugs that replicate the gene expression profile associated with EWSR1/FLI1 knockdown.113 Cytarabine was one of the most effective drugs in modulating the gene expression profile to emulate that of Ewing sarcoma cells subjected to EWSR1/FLI1 knockdown. The COG then conducted a phase 2 trial of cytarabine in patients with relapsed Ewing sarcoma, with no responses among 10 patients.114
Another novel agent, YK-4-279, was developed to interfere with the interaction of EWS-FLI1 protein and RNA helicase A, an important mediator of EWS-FLI1.115 This agent has shown preclinical activity against Ewing sarcoma models. Efforts to move this agent or agents with a similar mechanism of action in the clinic are ongoing.
A high-throughput screening study sought to identify existing agents that were able to reduce expression of EWS-FLI1 target genes.116 Mithramycin was the lead compound emerging from this screen. Follow-up experiments demonstrated preclinical activity against Ewing sarcoma models, leading to an ongoing clinical trial (NCT01610570).
Other novel approaches have tried to exploit additional vulnerabilities identified in laboratory studies of this disease. For example, a large body of literature has suggested that antiangiogenic strategies would be active against Ewing sarcoma.117 Based on these findings, several trials have been conducted. The COG conducted a trial using VDC/IE together with metronomic vinblastine and celecoxib in patients with newly diagnosed metastatic Ewing sarcoma.118 This regimen was tolerable, but outcomes did not appear different from previous outcomes reported for this population. A pilot study of bevacizumab added to vincristine, irinotecan, and temozolomide included two patients with Ewing sarcoma, both with objective responses.119 A nonrandomized multicenter trial of topotecan, cyclophosphamide, and bevacizumab for patients with recurrent Ewing sarcoma is ongoing (NCT01492673).
The insulin-like growth factor 1 receptor (IGF1R) has also been the subject of intense study in preclinical and clinical studies of Ewing sarcoma. The majority of Ewing sarcoma cells and tumor samples express IGF1R.120 EWSR1/FLI1 expression reduces the expression of IGF-binding protein 3, an endogenous inhibitor of IGF1R pathway activation.121 Preclinical studies of IGF1R small molecular inhibitors or inhibitory anti-IGF1R monoclonal antibodies have demonstrated robust activity against Ewing sarcoma models.122–125 In early phase clinical trials of IGF1R monoclonal antibodies, several patients with recurrent Ewing sarcoma demonstrated objective responses.126,127 Several phase 2 trials specifically for patients with recurrent Ewing sarcoma were developed, and response rates of approximately 10% were observed without significant single-agent toxicity.126,128–130 Based upon these promising results in the recurrent setting, the COG is developing a trial of an IGF1R monoclonal antibody together with VDC/IE for patients with newly diagnosed metastatic Ewing sarcoma.
The mammalian target of rapamycin has also been implicated as a potential therapeutic target in Ewing sarcoma. Preclinical studies indicate that rapamycin can reduce levels of EWS/FLI protein and also induce an EWSR1/FLI1-inactive gene expression signature.113,131 In vitro and in vivo studies have shown antitumor activity of rapamycin as a single agent and in combination with cytotoxic chemotherapy.132,133 To date, no trials of mammalian target of rapamycin inhibition specifically for patients with Ewing sarcoma have been conducted. However, in a phase 1 trial of ridaforolimus, one patient with advanced Ewing sarcoma had a confirmed partial response.134 One institution is conducting a trial in patients with high-risk Ewing sarcoma in which patients will receive a window of irinotecan, temozolimide, and temsirolimus prior to receiving interval-compressed chemotherapy (NCT01946529).
Most recently, laboratory studies suggest that Ewing sarcoma may be sensitive to inhibition of poly(adenosine diphosphate-ribose) polymerase (PARP). In a large-scale drug screen across myriad cancer histologies, Ewing sarcoma cell lines were found to be uniquely sensitive to the effects of a PARP inhibitor.135 Another study sought to investigate the mechanism of this observation.136 They noted that Ewing sarcoma cell lines have evidence of increased DNA damage, an effect that is potentiated in the presence of a PARP inhibitor. The use of a PARP inhibitor together with temozolomide resulted in dramatic preclinical responses compared to temozolomide alone or the PARP inhibitor alone. Based upon these observations, a phase 2 trial of olaparib has been conducted, with results pending (NCT01583543). Other trials of PARP inhibitors as single agents and in combination with other agents are in development.
Radiation Therapy for Patients with Newly Diagnosed Ewing Sarcoma
Advances in radiation delivery and target definition together with improvements in chemotherapy and surgery approaches technique have improved local control for patients with Ewing sarcoma. However, controversy has plagued decisions regarding local control measures particularly for pelvic primaries for which retrospective studies have indicated trends toward better local after surgery compared with radiotherapy, but failed to reach statistical significance. Such studies are fraught with selection bias as patients chosen for radiotherapy rather than resection tend to be those with larger tumors and less robust response to chemotherapy.137,138 Nonetheless, when feasible, surgery is generally the preferred local therapy modality and radiation is reserved for patients in whom anticipated morbidity from surgery is prohibitive. This leaves approximately 20% of children on COG studies receiving definitive radiation and 15% receiving adjuvant radiation for primary tumor control.
A prospective randomized trial comparing surgery and radiotherapy as local control modality for Ewing sarcoma at any primary site has never occurred and will never take place. Thus we are left with interpretation of data collected for cohorts in which local control decisions are made based on parameters including tumor location, size, response to chemotherapy, patient age, and patient preference based on expected morbidity of each procedure. One such informative study is an analysis of patients with pelvic Ewing sarcomas treated on INT-0091, a randomized controlled study that compared two chemotherapy regimens: VACA and VACA-IE.139 Treating physicians chose the local control approach, be it surgery, radiation, or both. The authors sought to assess local control modality after adjusting for tumor size and chemotherapy type. Of the 75 studied patients, 12 underwent surgery, 44 received radiotherapy, and 19 received both. Local control rates (80% to 90%) were quite promising even in this group with unfavorable primary site. The 5-year cumulative incidence of local failure was 21% with no significant differences by tumor size or local control modality. However, there was a trend toward benefit in local control with VACA-IE chemotherapy (11% versus 30%; p = 0.06). Of note, despite lack of statistical significance, combined surgery and radiotherapy was associated with the lowest rates of local relapse (10.5% for surgery + radiotherapy compared with 25% for either surgery or radiotherapy alone).139 Similar results, although again, not statistically significant, were reported for the entire cohort of patients, with 2% local recurrence for patients receiving combined modality treatment compared with 5% for surgery and 9% for radiotherapy alone (p = not significant).89 Not surprisingly, European studies have historically favored combined modality approaches for local control, with 63% of patients treated on EICESS 92 receiving combined surgery and radiation, 19% treated with surgery alone, and 18% with radiotherapy alone. Impressive local control rates were reported, with 95% when surgery was a component of local control compared with 75% when only radiotherapy was used.140 A dearth of reports describes the durability of local therapy modalities and the functional outcome of each approach. With a particular focus on cure rates, contemporary clinical trials have emphasized event-free survival and overall survival as end points, but better information regarding functional outcomes is critical to informed decisions regarding local therapy modality for individual patients.
Although postoperative radiation has gained favor for patients receiving combined modality therapy for local control, consideration should be given to preoperative radiation for a select group of patients with Ewing sarcoma. For these patients, the goal of preoperative radiotherapy is not to allow an inoperable tumor to become operable, as the data for this approach is lacking for Ewing sarcoma. Rather, in patients for whom the choice of combined modality therapy is made, the rationale for preoperative radiation lies primarily in reducing side effects. Preoperative radiation may allow for smaller fields, less normal tissue exposure, and perhaps lower doses, as has been reported for adults in a prospective, randomized study for adults with soft tissue sarcomas.141
Appropriate uses of radiotherapy for patients with newly diagnosed Ewing sarcoma include definitive radiation therapy for unresectable tumors, adjuvant radiation for tumors with incomplete surgical resection or intraoperative spill, chest wall tumors with ipsilateral pleural-based secondary tumor nodules or positive pleural fluid cytology, and pathologically involved lymph nodes. Patients with complete resections (R0) after neoadjuvant chemotherapy with a clear margin (defined as no viable tumor at the cut surface) should not receive radiotherapy; there are some subtle points to be highlighted. These guidelines incorporate not only the traditional factor of extent of surgical resection but also the parameter of percent of tumor necrosis more commonly used in European protocols. Thus, for resected tumors that have >90% necrosis, if the tissue at the margin is bland scar or loose fibrous tissue, margin status should be considered negative and no postoperative radiation should be administered. However, if inflammatory tissue or coagulative tumor necrosis is present at the margin (the cytoarchitecture of the tumor cells is preserved), postoperative radiotherapy is required. In contradistinction, for resected tumors with <90% necrosis, the cut surface of the resected tumor must be normal nonreactive tissue in order to be considered pathologically negative and radiotherapy avoided.
The role of preoperative radiotherapy must be carefully considered. Preoperative radiation should not be undertaken with the goal of rendering an inoperable tumor operable and should be considered only in resectable tumors in select sites such as pelvis and chest wall for which a higher risk of positive microscopic margins is present. Complete surgical excision should be undertaken within 2 weeks of 36 Gy of radiation. Standard radiotherapy doses are 50.4 Gy for microscopic residual and 55.8 Gy for gross residual disease.
One note of caution is in order for the administration of radiation to patients with Ewing sarcoma. High-dose chemotherapy with busulfan/melphalan is now often used for patients with metastatic disease. In late 2003, the pediatric oncology community was alerted to possible severe complications after busulfan/melphalan high-dose chemotherapy and irradiation of the spinal cord.142 And a subsequent amendment restricted the radiation dose to the spinal cord to 30 Gy after busulfan/melphalan high-dose therapy. To add to these concerns, in 2005, reports from France described severe gastrointestinal toxicities after busulfan/melphalan and pelvic radiotherapy. A detailed analysis of the European Ewing Tumor Working Initiative of National Groups Ewing Tumour Studies-99 study showed further serious toxicities with paraplegias and bowel obstructions leading to deaths.143 However, in follow-up studies by the German Pediatric Oncology and Hematology Society group, similar severe toxicities were not observed. Although the German group did not confirm such severe toxicities, it must be noted that whereas the median follow-up for gastrointestinal side effects was longer than the longest period for development of radiation associated bowel toxicities in France, the median follow-up for spine toxicities was only 7 months and therefore caution must be exercised. These differing findings may also be the result of lower doses and smaller volumes in the German Pediatric Oncology and Hematology Society group compared to those used in France. Given the critical role of radiotherapy for local control in many patients with Ewing sarcoma, if radiation is anticipated as part of a multimodality approach to local control in patients with axial tumor sites, consideration should be given to avoiding busulfan/melphalan high-dose therapy.144
Bone metastases in Ewing sarcoma receive the same dose of 55.8 Gy as does gross disease at the primary site. However, these metastatic sites are often treated at the completion of chemotherapy, months after local control measures, because protracted external beam radiation to such sites might include significant bone marrow volumes and ultimately compromise the ability to administer chemotherapy. Thus a role has emerged for hypofractionated stereotactic body radiotherapy (SBRT) for bone metastases. Safety of administration of such large doses to highly targeted volumes has been aided by improvements in immobilization and image-guidance techniques. Local control rates of 75% to 90% have been achieved by SBRT to spine metastases using 18 to 30 Gy in a single fraction or 24 to 60 Gy in five fractions.145 In the setting of metastatic bone sarcomas in children, the advantages of SBRT to bone metastases are particularly appealing because completion of the radiation course in one to five fractions with equivalent disease control rates, as conventional fractionation offers the added advantages of minimizing interruptions in systemic therapy and irradiation of smaller volumes.146
Systemic Therapies for Osteosarcoma
Chemotherapy for Patients with Newly Diagnosed Osteosarcoma
Chemotherapy plays little role in the management of patients with low-grade or surface osteosarcoma. Historically, there was controversy about the role of chemotherapy in the management of patients with high-grade osteosarcoma. In light of this controversy, the Multi-Institutional Osteosarcoma Study (MIOS) was conducted.147 Patients with newly diagnosed localized extremity high-grade osteosarcoma were randomized after complete surgical resection to observation or to adjuvant chemotherapy. Chemotherapy consisted of 45 weeks of combination therapy including bleomycin, cyclophosphamide, and actinomycin-D (BCD) cycles, high-dose methotrexate cycles targeted to achieve 1,000 micromolar peak concentration, and cisplatin/doxorubicin cycles. Thirty-six eligible patients were randomized. Of the 18 patients randomized to adjuvant chemotherapy, 6 (33.3%) recurred. Of the 18 patients randomized to observation, 15 (83.3%) recurred and 2 of the 3 patients in this group who did not recur did not accept the outcome of the randomization and were treated with adjuvant chemotherapy. At 2 years from randomization, 66% of the patients randomized to receive adjuvant chemotherapy were relapse-free compared with only 17% of the patients randomized to observation. These results confirmed the significant impact of chemotherapy on outcomes of high-grade osteosarcoma, and this trial established a new standard of care for this disease.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


