Marker
Structure/function
Ligand
Effect of activation
BDCA-2/BDCA-4
Associated with FcεRlγ to form a signaling receptor complex
ITAM
Upon ligation, they inhibit TLR activation and release of type I IFN
CD4
A glycoprotein expressed on the surface of T-helper cells, monocytes, macrophages, and dendritic cells
It recognizes the TCR-MHC class II complex and is required together with the CD3 zeta chain for the recognition of antigens
Activation of pDCs
CD 123
The IL-3 receptor (70KD) is composed of a ligand specific alpha subunit and a signal-transducing beta subunit shared by the receptors for interleukin 3 (IL3), colony-stimulating factor 2 (CSF2/GM-CSF), and interleukin 5 (IL-5)
IL-3
Amplification of inflammation
IL-T3
Characterized by its cytoplasmic ITIM domain
Fc receptor
Tolerance induction
IL-T7
Characterized by its cytoplasmic ITIM domain and is also expressed on B, T, and NK cells
IFN I
Inhibition of release of Type I IFN (negative feedback)
CD-11 c
A heterodimeric integral membrane protein composed of an alpha chain and a beta chain. It is present only on mouse, but not human, pDCs
ICAM-2 and VCAM-1
Induces cell activation; it is an adhesion receptor that is implicated in phagocytosis of latex beads and bacteria in the absence of complement. It plays an important role in the inflammatory response and can lead to the production of proinflammatory cytokines after an APC response
TLR-7
An intracellular endosomal pattern recognition receptor
Single-stranded RNA
Upregulation of CD40, CD80, CD86, and CCR7. Induction of high levels of Type I IFN. Does not induce IL-12p70 production
TLR-9
An intracellular endosomal pattern recognition receptor
Unmethylated CpG oligonucleotides from bacterial DNA
Upregulation of CD40, CD80, CD86, CD83, HLA-DR, and CCR7. Upregulation of Type I IFN, IL-6, TNFa, IL-8, and IP-10. Does not induce IL-10 secretion
In addition, recent evidence demonstrated that CD9+ Siglec-Hlow pDCs secrete IFN-α when stimulated with TLR agonists, induce CTLs, and promote protective antitumor immunity. By contrast, CD9neg Siglec-Hhigh pDCs secrete negligible amounts of IFN-α, induce Foxp3+ CD4+ T cells, and fail to promote antitumor immunity [36]. Although newly formed pDCs in the bone marrow are CD9+ and are capable of producing IFN-α after aggregating in peripheral tissues, they lose CD9 expression and the ability to produce IFN-α. Therefore, recognition of the pDC surface markers is actually very important not only to distinguish pDCs from mDCs and other cell types but also to identify their function and to allow researchers to isolate them. To date, Bdca2-DTR [37] and Siglec-H-DTR models [38] are the recently developed appropriate murine models used to study the role of pDCs in the pathogenesis of various diseases. These mouse models allow the study of pDCs in pathophysiological conditions through the depletion of pDCs by diphtheria toxin (DT) using the human diphtheria toxin receptor (DTR) that is driven by the BDCA2 promoter, as the mouse receptor for DTR binds several orders of magnitude more weakly to DT. However, many studies have also been conducted by using specific depleting antibodies (Abs), such as 120G8 Ab [39], BST-2 Ab [40], and mPDCA-1 [41] in vivo. All these Abs bind to the same surface marker (BST-2 or CD317). Ab-depletion models seem to be less specific than DTR models, but still very efficient in pDC depletion, thus allowing the investigation of the role of pDCs during steady state and pathological conditions. The limitation of Ab-mediated pDC depletion stands on the role of some molecules, such as BST-2, which is also expressed by stromal and other immune cells after an inflammatory stimulus [40].
11.4 Activation of pDCs
Plasmacytoid dendritic cells are highly specialized at sensing nucleic acids via the intracellular pattern recognition receptors, Toll-like receptors (TLR) 7, and TLR9 [16, 34]. pDCs and mDCs have a different repertoire of TLR expression [16, 18, 34]. Human and mouse mDCs can express TLR1, TLR2, TLR4, TLR5, TLR7, and TLR8, while pDCs selectively express high levels of TLR7/TLR8 and TLR9 [42]. TLRs are a family of receptors associated with the innate immune response [43]. In particular, TLR7 recognizes single-stranded RNA enriched with guanosine or uridine from viruses, synthetic imidazoquinolines, and guanosine analogs [43]. On the other hand, TLR9 is activated by unmethylated CpG oligodeoxynucleotide (CpG-ODN) motifs typical of viruses and bacteria [43]. Interestingly, the response of human pDCs is dependent upon the class of synthetic CpG-ODN used to stimulate them. Stimulation with CpG-A (D)/2216 ODN induces sustained high IFN-α production by pDCs, but minimal upregulation of cell surface maturation markers including CD80, CD86, and major histocompatibility complex class II (MHC-II) [44, 45] has no effect on B cells (which also express TLR9). On the other hand, stimulation with CpG-B (K)/2006, a strong B-cell activator, results in increased expression of costimulatory and Ag-presenting molecules and higher IL-8 and TNF-α secretion, but lower levels of IFN-α production by pDCs. Two distinct pathways of IFN-α/IFN-β production have been identified regarding stimulation with CpG-A vs. CpG-B [45]. pDCs constitutively express IRF-7 and synthesize high levels of IFN-α in response to CpG-A, which also triggers an autocrine feedback loop involving the IFN receptor-dependent pathway [42]. In contrast, IFN-α/IFN-β induction by CpG-B is independent of the IFN-α/IFN-β receptor loop [45, 46]. Recently, CpG-C, a new class of CpG ODN in which structural elements of CpG-A and CpG-B have been combined, has emerged. This sequence activates B cells and induces IFN-α production by pDCs [47]. Furthermore, non-CpG-containing ODNs have been shown to bind human TLR9 [47, 48] and to stimulate pDCs [49].
TLR7 and TLR9 are very sensitive to different stimuli; the first triggers ssRNA viruses and the latter responds to DNA viruses [50]. TLR7 and TLR9 activation recruits a cytoplasmic adaptor, myeloid differentiation primary response gene 88 (MyD88), which is able to assemble a multiprotein signal-transducing complex-inducing interferon regulatory-factor 7 (IRF-7) activation [43]. MyD88 also leads to TRAF-6-mediated NF-κB and MAP-kinases (MAPKs) activation, essential for the transcription of proinflammatory cytokines, chemokines, and costimulatory molecules [43, 51].
The exposure of pDCs to TLR7 or TLR9 ligands can lead to the production of type I IFN and proinflammatory cytokines, such as TNF-α, and chemokines, such as IL-8 (CXCL8) [1, 16, 18]. Constitutive expression of IRF7, which is different from mDCs in which induction is needed, renders pDCs high producers of type I IFN [1, 16, 18], regulating T-cell immunity, leading toward a Th1 and cytotoxic T lymphocyte polarization and activation of mDCs, NK cells, and B cells [1, 16, 18]. Remarkably, IFN-α modulates several aspects of the immune system, including pDC survival [52], mDC differentiation, modulation of Th1 and CD8+ T-cell responses, cross-presentation, upregulation of MHC and costimulatory molecules, activation of NK cells, and induction of primary Ab responses [53]. However, a recent study found that type I IFN negatively controls pDC turnover in that an overproduction of type I IFNs can lead to the death of pDCs during steady-state conditions and viral infections [50]. pDC activation can also lead to the production of IL-12p70, IL-1β, and IL-6 [54]. Furthermore, recent discovery found that pDCs may mediate the release of IL-10 [26]; however, another group [55] showed that these cells do not directly produce IL-10 (Fig. 11.1).
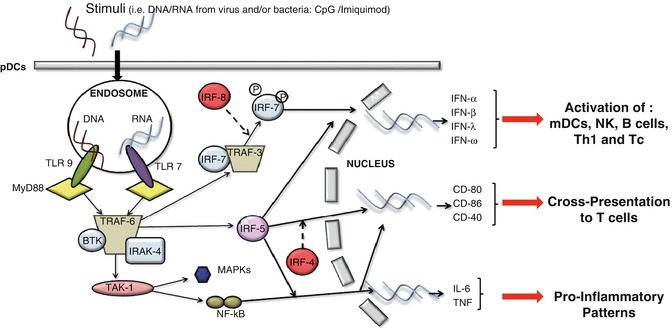
Fig. 11.1
The recognition of stimuli, such as DNA or RNA motifs from viruses and bacteria, by pDCs via TLR7 and/or TLR9, induces the activation of MyD88-dependent signalling pathways that lead to the expression of cytokines such as IL-6 and TNF-α, costimulatory molecules such as CD80, and the synthesis/release of type I IFN
Moreover, it was recently demonstrated that pDCs produce high amounts of granzyme B [56], which is effective only in combination with perforins mainly produced by cytotoxic T lymphocytes (CTLs). This further connects pDCs to adaptive immunity. Additionally, in the absence of an “efficient” adaptive CTL immunity, pDCs can behave as killing DCs due to the release of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and to the induction of DR5 expression, a TRAIL receptor, on the cell target [37, 56].
A diversity of C-type lectin receptors (CLRs) has been identified on DC subsets, including DC-SIGN (CD209), DEC-205 (CD205), langerin (CD207), mannose receptor (CD206), BDCA-2, and dectin-1. CLRs typically recognize carbohydrate-rich structures on microbes and self-antigens [35]. They have been implicated in cell adhesion and regulation of signaling events (e.g., BDCA-2), migration and homing (e.g., DC-SIGN), Ag uptake and processing for MHC-II presentation to T cells (e.g., DC-SIGN, BDCA-2, langerin, and mannose receptor), cell-cell transmission of pathogens (e.g., DC-SIGN), and tolerance to self-antigens (e.g., DEC-205). pDCs express BDCA-2 and BDCA-4, dectin-1, and possibly DEC-205 but lack DC-SIGN and langerin, found on CD34+ and monocyte-derived DCs and Langerhans cells (LCs), respectively [57]. The physiologic function of CLRs on pDCs remains unknown. Anti-BDCA-2 Abs are rapidly internalized and efficiently presented to T cells, suggesting a role in Ag capture and presentation [35]. Interesting relationships between CLRs and TLRs have been documented. In mDCs, interaction of DC-SIGN with lipoarabinomannan secreted by mycobacteria inhibits lipopolysaccharide (LPS)-induced DC activation through TLR4 [58]. This mechanism may permit pathogens to evade immune responses and perpetuate tolerance to self-antigens in the face of TLR activation by microbes. On the other hand, it has been shown that dectin-1 collaborates with TLR2 in inducing proinflammatory cytokine secretion in murine macrophages and DCs [59]. Whether BDCA-2 has any connection to TLRs in pDCs remains to be elucidated. However, early reports have shown that secretion of type I IFNs by pDCs in response to influenza virus (most likely triggering TLR7/8) or to complexes of plasmid DNA and anti-DNA Abs (possibly stimulating both FcR and TLR9) is significantly inhibited by ligation of BDCA-2 with anti-BDCA-2 Ab [35]. It is worth noting that BDCA-2 is downregulated after pDCs maturation and that mature pDCs secrete less IFN-α/IFN-β in response to viral stimuli than immature pDCs do [60, 61]. BDCA-2 has an intracellular domain of 21 amino acids without known motifs implicated in signal transduction; however, ligation induces Src family protein-tyrosine kinase-dependent intracellular calcium mobilization and protein-tyrosine phosphorylation of intracellular proteins [35]. BDCA-4 (neuropilin-1) is also upregulated in blood mDCs after overnight culture and may participate in DC-lymphocyte interactions [62].
11.5 pDCs: Bridging the Gap Between Innate and Adaptive Immunity
The production of type I IFNs by pDCs represents the bridge between the innate and adaptive immune system. Type I IFN (IFN-α and IFN-β) is an important component of the innate immunity, especially during viral infections [16, 18]. In contrast to mDCs, pDCs produce high amounts of type I IFNs upon activation [16, 18], which both amplify its own production in an autocrine manner and induce the release of other proinflammatory cytokines such as IL-12p70 from mDCs and NK cells [63] (Fig. 11.2a). Activation of mDCs diverts the immune environment toward a Th1-like bias, during which IFN-γ production facilitates Th1 differentiation [16, 18, 63], long-term T-cell immunity [18, 63], and a CTL-mediated response [64], as well as proliferation and survival of T cells [63, 64]. Moreover, through the production of IL-6 and type I IFNs, pDCs induce B cells to differentiate into plasma cells which are immunoglobulin (preferentially IgG and IgM)-producing cells (Fig. 11.2b). In the process of B-cell activation, a key role is played by the CD70 receptor expressed on pDCs, as it can induce the differentiation and the proliferation of IgG-producing B cells [65] (Fig. 11.2b).
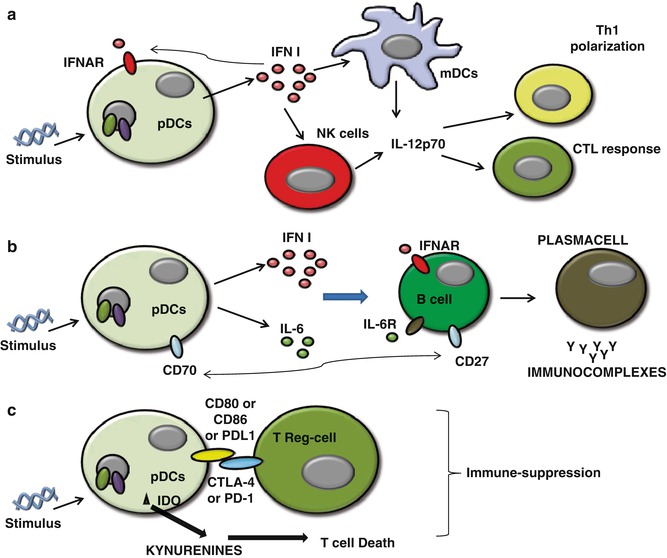
Fig. 11.2
(a) Activated pDCs produce high amounts of Type I IFNs which both amplify its own production in an autocrine manner via the expression of IFNAR on themselves and induce the release of other proinflammatory cytokines such as IL-12p70 from mDCs and NK cells that lead to Th1 and CTL polarization; (b) pDCs induce B cells to differentiate into plasma cells via the activation of IFNAR, IL-6R activation, and the interaction of CD70-CD27 on B cells; (c) pDCs can lead to immunosuppression via both direct interaction with Treg (CD80 or CD86+CTLA-4 or PD-L1+PD1) and the release of IDO-induced kynurenines metabolites which induce Th1 cell toward death
In addition, activated pDCs can undergo other important phenotypic changes that induce them to change their phenotype toward an mDC phenotype [1]. The upregulation of MHC and T-cell costimulatory molecules enables pDCs to engage and activate naïve T cells [66–68]. There have been many controversies regarding the role of pDCs to prime T cells and cross-present Ags [68]. The expression of MHC and T-cell costimulatory molecules is not as high as in mDCs, and this is why pDCs are less efficient than mDCs at priming T cells [69]. Moreover, the repertoire of Ags that can be presented by pDC-derived MHC molecules is more restricted than those of mDCs because not all of these Ags reach the endocytic compartment into pDCs [68, 69]. However, some pDC receptors such as BDCA2, Siglec-H, and DCIR are able to bind Ags, mediate endocytosis, and process and present to T cells [68, 69].
Interestingly, activated pDCs can also promote Th2-like immune responses [63] underlining their functional plasticity. There is evidence that IFN-α stimulates the differentiation of pDCs into Th1-polarizing pDCs, whereas in the absence of IFN-α but only in the presence of proinflammatory signals, pDCs can also stimulate Th2 polarization/differentiation [70]. Moreover, some authors reported that CpG-activated pDCs exert a strong immunosuppression and induce the differentiation of allogeneic CD4+CD25+ T cells into CD4+CD25+ regulatory T cells in tumor conditions [50, 55]. Very interestingly, pDCs can directly or indirectly recruit Treg cells via PD-L1/PD-1 axis [71] (Fig. 11.2c), release of immunosuppressive cytokines, such as IL-10 [55, 71], and the membrane tolerogenic inducible costimulator ligand (ICOS-L) [72].
pDCs can also synthesize large amounts of functional indoleamine 2,3-dioxygenase (IDO), which requires autocrine release of type I IFN, upon TLR9 and CD200R ligands’ stimulation [16]. IDO-derived metabolites promote T-cell death [55, 73] and suppresses T-cell immunity in normal and pathological settings. In the same manner, reduced tryptophan amounts can lead to the release of regulatory cytokines, such as IL-10 [74], associated with a tolerogenic environment.
Taken together as a whole, these data suggest that pDCs represent a key effector cell in both innate and adaptive immunity regulation [1].
11.6 pDCs and Human Diseases
A wide spectrum of human diseases including infection, autoimmunity, and cancer are associated with accumulation of pDCs in lymphoid and peripheral tissues strictly correlated to the reduction of these cells in the peripheral blood [21]. For many of these diseases, compelling evidence supports a pathogenic role of pDCs, mainly related to either the increase or reduction of proinflammatory or antiinflammatory functions of pDCs. Alternatively, pDC accumulation might exert an adjuvant immune function, as in viral infection, and in imiquimod-treated cancers, where they seem to encounter an antiviral and antitumor activity. In many other pathologies, information available is still limited, and pDC biology is largely unknown.
11.6.1 Role of pDCs in Human Infections
pDCs have been most extensively studied during HIV and chronic viral hepatitis, particularly hepatitis C virus (HCV) infections. The emerging picture suggests an important role for pDCs in these infections; however, the exact mechanism and consequences of pDC activity are controversial at present [75]. pDCs can respond to HCV and particularly to HCV-infected hepatocytes which induce pDCs to signal via an endocytosis- and IRF7-dependent mechanism, but not via the NF-κB pathway, implying a non-full functional response of pDCs that contribute to the evasion of immune responses by HCV [76]. In contrast, other studies demonstrated normal pDC functionality in chronic HCV infection [77]. The resolution of this controversy would establish pDCs either as a weak link of anti-HCV immune response or as a potentially powerful effector type that can be harnessed for immunotherapy of chronic HCV.
Similarly, pDC dichotomy is observed in HIV infection, in which some authors assume that pDCs can be infected with the HIV and/or respond to it with robust IFN secretion [78], while others reported impaired activity of pDCs in HIV infected patients [79, 80]. Interestingly, pDCs are progressively depleted from the blood of infected patients, either through infection-induced death or due to redistribution to lymphoid organs. The key unresolved question is whether HIV-induced pDC activation is beneficial or harmful for the host. On one hand, IFN secretion by pDCs was shown to inhibit viral replication in T cells and promote pDC and cDC maturation, leading to the killing of infected T cells. In this context, it is likely that HIV may have evolved mechanisms to suppress pDC activation, e.g., through BDCA-2 ligation [81], which disables pDC functions as APCs and type I IFN-producing cells. On the other hand, the same functions of pDCs may exacerbate T-cell depletion, e.g., by disseminating HIV to uninfected CD4+ T cells or by bystander T-cell killing. Most importantly, elevated IFN response by pDCs may contribute to chronic immune activation and faster T-cell depletion [82]. It is plausible that the function of pDCs in HIV infection changes from protective to pathogenic as the disease progresses. At the early stages of infection, IFN production and virus cross-presentation by pDCs may help limit virus spread and mount cytotoxic T lymphocyte responses; whereas as the virus replication escapes control, IFN secretion may drive polyclonal T-cell hyperactivation and depletion [77]. The eventual loss, redistribution, or functional impairment of pDCs at the late stages of infection would contribute to immunodeficiency. Thus, the role of pDCs in HIV and HCV infections highlights the power and the danger of pDC activation and reveals another strategy of immune system subversion by these viruses.
11.6.2 Role of pDCs in Autoimmune Diseases
Several autoimmune diseases are associated with elevated levels of type I IFNs, implying a potential role for pDCs in cytokine production [83]. To date, the strongest evidence for pDC involvement has been accumulated from the study of two diseases: psoriasis and systemic lupus erythematosus (SLE) [84]. In psoriasis, early skin lesions are highly infiltrated by activated pDCs, corresponding with decreased numbers of circulating pDCs [85]. Blocking IFN production by pDCs using anti-BDCA-2 Ab inhibited the development of skin lesions in a xenograft mouse model, providing causal proof of pDC function in the disease [85]. Gilliet’s group [86] identified the activating stimulus for pDCs as complexes of self-DNA with the antimicrobial peptide LL-37. This and possibly other homologous proteins promote the aggregation of released cellular DNA and RNA into large complexes that efficiently activate pDCs [86, 87]. Although the origin of these immunostimulatory complexes and the consequences of pDC activation remain to be elucidated, the major role of pDCs in psoriasis is well established. Similarly, lupus patients show a decrease in circulating pDCs and the accumulation of activated, IFN-producing pDCs in affected tissues such as the skin [88]. The hallmark of lupus is the production of antinuclear Abs and immune complexes of such Abs with endogenous nucleic acids were shown to activate pDCs through TLR7/9 [89, 90]. These complexes may be delivered into the endosomal compartment of pDCs via Fc receptor II (FcγRII) [89, 91], and their stimulatory capacity can be augmented by the nuclear DNA-binding protein HMGB1 [92]. In addition, self-DNA forms complexes with LL-37 and other antimicrobial peptides released by neutrophils, and the resulting complexes induce IFN secretion in pDCs through TLR9 [92]. Notably, TLR-activated pDCs become resistant to glucocorticoids, which could underlie the limited efficacy of these drugs in lupus [93, 94]. The direct causal relationship between pDC-derived IFN and lupus progression/severity is hard to establish in the human system and should await for elucidation in animal models. Nevertheless, the likely connection between the formation of nucleic acid-containing immune complexes, pDC activation, and IFN secretion and the pronounced IFN signature of the disease makes a strong case for the pDC as a major player in lupus pathogenesis [77]. Overall, the aberrant conversion of self-nucleic acids into ligands for TLR7/TLR9 on pDCs (via immune complex formation, antimicrobial peptide binding, and other mechanisms to be discovered) may represent a common pathogenesis step in psoriasis, lupus, and possibly other autoimmune diseases such as Sjögren’s syndrome [95].
The activity of pDCs in viral and autoimmune diseases might teach us how and why pDCs highly populate cancerous masses playing a pivotal role for the tumor immune microenvironment.
11.6.3 Role of pDCs in Cancer
Recent studies have shown that the density and location of immune cells in primary tumors can predict patient survival [96], supporting the notion that monitoring local immune response might represent a critical step in predicting patient prognosis and likely the response to antitumor strategies [97]. pDCs have been found in a variety of neoplasms; nonetheless their function is still unknown. Solid tumors, such as head and neck, breast, ovarian, lung cancer, and skin tumors, are populated by non-active pDCs [97]. Clinical studies have suggested a direct correlation between reduced numbers of circulating pDCs and higher presence of these cells into malignant masses [1, 97]. Although the causal relationship is still under investigation, recent results from mouse models are starting to define the specific role(s) of pDCs in tumor masses. The mechanism that induces the recruitment of pDCs to the tumor site is not clear. Circulating pDCs express multiple chemotactic receptors such as CXCR4 and ChemR23 being the only biological active receptors in healthy donors [28]. CXCR4 binds CXCL12, widely expressed in tissues and which most likely represents the main axis for pDC accumulation in human tumors [25]. CXCL9, CXCL10, and CXCL11, which bind CXCR3, present on pDCs, are all IFN-inducible proteins and might be involved in pDC infiltration [98]. In addition, cytokines such as CXCL10, CXCL12, and chemokines, such as CCL2, are released by tumor and stromal tumor-associated cells, such as cancer-associated fibroblasts (CAFs), allowing pDCs to migrate from the circulation to the injured tissue [23]. Accordingly, Drobits et al. demonstrated that CCL2 produced in the inflamed skin of tumor-bearing mice facilitated pDC recruitment [56].
Once recruited, pDCs seem to be important players in cancer immunoediting as their capacity to bring together the innate and the adaptive immunity. In particular, it seems that critical role is played by type I IFNs. Endogenously produced IFN-α/IFN-β was required for the prevention of the growth of primary carcinogen-induced sarcoma [99]. In this study, host hematopoietic cells were critical targets of IFN-α/IFN-β during the development of protective antitumor responses [99]. pDCs have been widely described as professional type I IFN-producing cells; therefore, the higher presence of pDCs in the tumor mass might directly link pDCs to cancer immunoediting in that pDCs may behave as antitumor cells. However, other reports showed opposite activities of pDCs in cancer. Animal studies demonstrated that tumor-associated pDCs (TApDCs) are defective in type I IFN production but instead secrete immunosuppressive factors responsible for tumor progression [100, 101]. Similar to what described for viral infections and autoimmune diseases, the dichotomy of pDCs in cancer might underlie their phenotype and maturation state.
11.6.3.1 Antitumor Activity of pDCs
Type I IFNs are pleiotropic cytokines with a demonstrated clinical benefit to cancer patients and have recently emerged as the connection bridge between tumor cells and the immune system [102]. pDCs produce large amounts of type I IFNs upon TLR7 and TLR9 stimulation. Drobits et al. showed that the intratumoral stimulation of pDCs with imiquimod renders these cells cytotoxic and contributes to tumor regression independently from conventional adaptive immune mechanisms, but via the production of TRAIL and granzyme B secretion by pDCs via IFNAR1 signaling [56]. However, the role of TApDC-derived granzyme B in the absence of perforins not produced by pDCs still remains to be elucidated.
Another mechanism that may underlie the antitumor activity of TApDCs is their antigen-presenting activity. Although in their immature state, TApDC are still capable to internalize Ags in vivo and to activate CD4+ T cells [103]. The immature state of pDCs is reflected in that they have altered cytokine production in response to TLR-9 ligands in vitro, while preserving unaltered response to TLR7 ligands [104], which instead seem to have potential antitumor activity. To date, imiquimod is in phase III clinical trial against melanoma. In contrast to these results, systemic administration of CpG favored pDC-induced lung tumor progression [105], as also observed in a mouse model of breast cancer [104]. Similar to the data showed by Drobits et al., Mercier et al. proved that, although CpG did not alter TApDC activity, the intratumoral administration of a TLR7 ligand led to TApDC activation and displayed a potent curative effect in a type I IFN-dependent manner [56]. In addition, Liu et al. [106] demonstrated that the intratumoral activation of pDCs via CpG could induce NK cell-dependent tumor regression in a melanoma animal model. Remarkable is that TLR9 expression and responsiveness is impaired by tumor-derived components [107]. ILT7 on pDCs binds BST-2 expressed by tumor cells and their interaction inhibits type I IFN production by pDCs, disabling TLR9-dependent signaling pathways [108]. Moreover, tumor-derived TGF-β and TNF-α have been identified as the main in vivo mechanisms blocking type I IFN production by pDC in tumors through inhibition of IRF7 signaling complex, leading to a negative impact of defective pDCs in breast cancer through Treg expansion [109].
Taken altogether, these data supported the rationale to use TLR7 ligands to restore TApDC activation in both breast and skin cancer. However, it still remains to be determined how the activation of TLR7 and TLR9, which is MyD88-dependent, on pDCs, can behave differently according to the tissue specificity and on the route of administration.
11.6.3.2 Pro-tumor Activity of pDCs
Several evidence have shown the prevailing immunosuppressive activity of pDCs due to both of the impairment in type I IFN production and the release of pro-tumor factors [1]. Stimulation of lung tumor-bearing mice with systemic CpG, a TLR9 ligand, did not lead to the same results as observed by Liu et al. [106]. Activation of pDCs through CpG had the opposite effect in that pDC activation increased the recruitment of Tregs and limited the inflammatory cell influx to the lung, thereby establishing an immunosuppressive environment enabling tumor growth [1, 105, 109]. The same was observed in another mouse model of breast cancer in which in vivo depletion of pDCs delayed tumor growth showing that TApDC provide an immune-subversive environment, most likely through Treg activation thus favoring breast tumor progression [110]. The discrepancy in these data and the one from Liu et al. [106] could be a result of tissue-specificity and route of CpG administration which is very important in determining the tumor microenvironment, which in turn strongly influences immune cell phenotype. Moreover, in the absence of a specific stimulus, pDCs in the tumor mass have been associated with the development and maintenance of the immunosuppressive microenvironment [111]. Similar to mice, human pDCs in tumor masses are in their immature phenotype; nonetheless, a thorough study has never been conducted on the role of these cells in human tumor microenvironment. Nevertheless, it is clear that pDCs play a fundamental role in the tumor microenvironment. The specific depletion of pDCs induced lung tumor regression with a concomitant Th1 polarization that arrested tumor progression [105]. On the other hand, stimulation of TLR7, rather than TLR9, can subvert the immunosuppressive activity of TApDCs. TLR7-dependent pathway induced melanoma regression in mice [56] through the transformation of pDCs into tumor-killing cells able to produce granzyme B and TRAIL. Likewise, another group revealed that human pDCs can kill melanoma cells in vitro under imiquimod and IFN-α stimulation [112]. While pDCs can produce high levels of granzyme B, their role as cytotoxic immune cells remains to be determined as they lack the pore-forming perforin [112]. On the other hand, it has been proposed that under IL-3 and IL-10 exposure, pDCs release abundant granzyme B, which in turn is capable of blocking T-cell proliferation, thus suggesting a new potential mechanism for tumor-immune evasion [112].
Several mechanisms have been postulated for the immunosuppressive nature of tumor-associated pDCs: (1) release of tolerogenic factors, (2) ILT-7 expression, (3) PD-L1 expression, (4) Siglec-H activity, and (5) induction of a Th2-like environment. Tolerogenic factors produced by tumor cells, such as PGE2 [113] and TGF-β [109], can alter type I IFN signaling pathway. Tumor-derived PGE2 and TGF-β act synergistically to block IFN-α and TNF-α secretion by pDCs [16, 109]. Opposite to IFN-α and TNF-α, IL-6 and IL-8 production are enhanced in PGE2- and TGF-β-treated pDC [114]. Both IL-6 and IL-8 promote immune-cell survival and chemotaxis but also enhance tumor cell proliferation and angiogenesis [115, 116]. Moreover, PGE2 is crucial for the secretion of other immunomodulatory factors such as SDF-1, the ligand for CXCR4, which is upregulated on both human pDCs and tumor environment [117]. Thus, pDCs can be retained in the tumor tissue via PGE2-induced sensitization for SDF-1 [29]. In further support, PGE2- and TGF-β-mediated retention of pDCs in the tumor tissue is accompanied by the suppression of the lymph node-homing receptor, CCR7 [113]. PGE2-exposed pDCs are unlikely to present Ags and to prime T cells in the regional LNs. Concomitantly, suppression of CD40 expression and the overexpression of CD80/86 on pDCs enhances and even promotes Treg activation via the negative regulatory receptor cytotoxic T-lymphocyte antigen-4 (CTLA-4) [118, 119] (Fig. 11.2c).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


