Fig. 1
Differentiation-dependent HPV Life Cycle. The HPV life cycle is intimately associated with the differentiation program of epithelial cells. HPV infects actively dividing cells in basal layer of the epithelium via a micro-abrasion. Following entry, viral gene expression is activated and episomal HPV DNA is maintained at approximately 50–100 copies per cell. HPV oncoproteins then enable the infected cells that exit the basal layer to remain active in the cell cycle. Once the infected cells begin to differentiate, the late promoter is activated, which results in the onset of the productive phase of the viral life cycle. During this phase, viral DNA amplification occurs and viral protein expression is increased. Finally, synthesis of viral capsids and packaging occurs in the uppermost differentiated layer of the epithelium, followed by release of the progeny virions
Upon infection, the virus establishes its double-stranded DNA genome in the nuclei of infected host cells (Howley and Lowy 2007). HPV gains entry to cells in the basal layer of the epithelium that become exposed through micro-abrasions (Moody and Laimins 2010). Infection of the basal layer allows the virus to establish a persistent infection, as the basal cells are the only cells of the epithelium undergoing active replication (Moody and Laimins 2010). Since the HPV genomes are only 8 kb in size, they do not encode viral polymerases or other enzymes required for viral replication. The virus must therefore rely on host cell replication machinery to facilitate viral DNA synthesis (Moody and Laimins 2010). Following entry, viral genomes are established in the nucleus as extrachromosomal plasmids, or episomes. In the infected basal cells, early viral gene expression is activated, and genome copy numbers are maintained at approximately 20–100 copies per cell (Moody and Laimins 2010). As HPV-infected basal cells divide, one of the infected daughter cells remains in the basal layer. The other daughter cell migrates away from the basal layer and begins to differentiate, resulting in the activation of the late viral promoter. This results in the onset of the productive phase of the life cycle, which includes viral DNA amplification, with copy number increasing to over 100 copies of HPV DNA per cell, and the onset of capsid gene expression. Finally, synthesis of viral capsids and packaging of viral genomes occur in the uppermost differentiated layer of the epithelium, ultimately resulting in the release of the progeny virions.
The signals that control the induction of late viral events in the life cycle are not well characterized, but studies have shown that HPV oncoproteins enable infected cells in the suprabasal layer to remain active in the cell cycle and to reenter S phase or arrest in G2/M to allow for viral amplification. This alteration of cell cycle control is essential for activation of the productive phase of the life (Moody and Laimins 2010). Furthermore, studies have indicated that viral proteins E6, E7, E1^E4, and E5 are needed for this activation, and these activities will be briefly summarized below.
1.2 The HPV Genome
The small, double-stranded DNA genome of all HPVs is approximately 8 kb in size. On average, HPVs encode eight major ORFs which are expressed from polycistronic mRNAs transcribed from a single DNA strand (Howley and Lowy 2007) (Fig. 2). The early proteins, E1, E2, E6, and E7, are expressed early in infection in undifferentiated cells and have drastically different functions. Sequences within the upstream regulatory region (URR) located in the non-coding region of the genome are responsible for regulation of viral transcription and replication. Expression of HPV gene products is directed from two different promoters, the early promoter and the late promoter (Moody and Laimins 2010). The early promoter, termed P97 in HPV 31, is located upstream of the E6 ORF and directs expression of early (E) gene products in undifferentiated cells. Early proteins include E1, E2, E6, E7, E1^E4, and E5. Translation of HPV messages occurs by a leaky scanning mechanism resulting in high levels of E6 and E7, but low levels of E1^E4 and E5 protein synthesis. The E1 and E2 proteins function in replication and transcription control, while E1^E4 modulates late viral functions. The late promoter, P742 in HPV 31, directs the expression of late (L) gene products and is located within the E7 ORF. Importantly, P742 is activated upon epithelial cell differentiation. Late proteins include L1 and L2 as well as E1^E4, and E5, and these are all expressed from P742 (Moody and Laimins 2010).
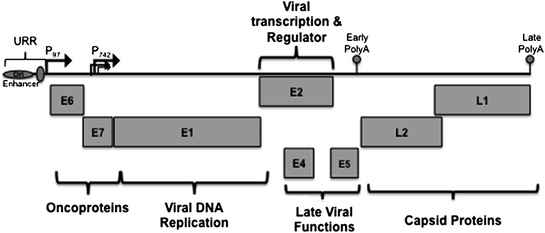
Fig. 2
Linear arrangement of HPV genome. The HPV genome is represented in linear form here for simplicity. The genomes are small, circular, double-stranded DNA genomes of 8 kilobases (kb) in size. There are on average 8 open reading frames (ORFs) (E1, E2, E4, E5, E6, E7, L1, and L2) expressed from a single polycistronic transcript transcribed from a single strand of DNA. The upstream regulatory region (URR) is located in the non-coding region and contains sequences responsible for regulating viral transcription and replication. Three general groups of HPV genes that are regulated during differentiation are the virus early promoter (P 97 ), the differentiation-dependent late promoter (P 742 ), and two polyadenylation signals (PolyA). P 97 is located upstream of the E6 ORF and directs expression of early genes in undifferentiated cells. P 742 is located within the E7 ORF, is activated upon differentiation of the host cells, and directs expression of late gene products. E6 and E7 are oncogenes involved in replication competence. E1 and E2 are genes involved in viral DNA replication and regulation of viral transcription. E4 and E5 are genes involved in late functions. L1 and L2 are the capsid proteins
1.3 Oncoproteins: E5, E6, and E7
The E6 and E7 proteins are expressed upon initial HPV infection of host keratinocytes, while E5 is primarily expressed in the late phase of the life cycle. Although these proteins all contribute to promoting tumor growth in host cells, each of these proteins has distinct functions. As previously discussed, E1 and E2 are responsible for replication and regulation of viral transcription, whereas E6 and E7 proteins are largely responsible for modulating cell cycle progression. In the HR-HPVs, E6 and E7 act as oncoproteins that are necessary for the development of genital carcinomas. Conversely, no such function has been demonstrated for LR-HPV proteins. While both E6 and E7 proteins are localized to the host nucleus, the E6 proteins are also detected in the cytoplasm of HPV-infected cells (Howley and Lowy 2007; Moody and Laimins 2010). Studies have shown that expression of E6 proteins is sufficient for immortalization of human mammary epithelial cells and transformation of NIH3T3 fibroblasts; however, expression of both E6 and E7 proteins is required for efficient immortalization of human keratinocytes (Howley and Lowy 2007).
E6 proteins are approximately 150 amino acids (18 kilodaltons) in size and contain two zinc-binding domains consisting of four Cys–X–X–Cys motifs (Howley and Lowy 2007). Studies have identified various E6-mediated activities that are mediated by interactions with over a dozen different proteins. One of the well-characterized interactions is the binding of E6 proteins to the tumor suppressor protein p53, affecting p53-dependent cell cycle regulation. The p53 protein is important in regulating the G1/S and G2/M cell cycle checkpoints following DNA damage (Slee et al. 2004; Oren 2003). Studies have shown that E6 proteins form complexes with an E3 cellular ubiquitin ligase, E6-associated protein (E6AP), and p53, resulting in rapid proteasomal (26S) degradation of p53 (Scheffner et al. 1990). In a normal response to DNA damage or unscheduled induction of replication, p53 is activated via various modifications. The activation of this short-lived transcription factor results in modulation of the cell cycle and in, some cases, activation of apoptotic processes. Activated p53 forms a homotetramer that transcriptionally activates expression of cell cycle regulatory proteins, such as cyclin kinase inhibitor p21, which is responsible for inducing a G1/S arrest (Ko and Prives 1996). The activation of p53 can also induce programmed cell death (apoptosis). Cell cycle arrest allows the cell to repair the damage to the DNA prior to entry to S phase. In the event that the damage is too extensive, the cell triggers apoptosis to prevent a cell from replicating damaged DNA. In addition, p53 is activated following viral infection. Given that HPV relies on host cell machinery and S phase entry to replicate its genome, the virus has devised a mechanism to disrupt normal p53 action. The E6-mediated proteasomal degradation of p53 results in the deregulation of the cell cycle, which allows the virus to persist and replicate its genome (Moody and Laimins 2010).
An additional HR E6 activity is its ability to interact with p300/CBP, a p53 co-activator (Patel et al. 1999; Zimmermann et al. 1999). The p300/CBP/E6 interaction prevents acetylation of p53, which down-regulates p53 activity, therefore blocking cell cycle arrest. The binding of p300/CBP occurs independently of E6-mediated degradation of p53 (Patel et al. 1999; Zimmermann et al. 1999). Interestingly, studies indicate that immortalization competency is not exclusively linked to p53-dependent mechanisms since E6 mutants incapable of degrading p53 are still able to immortalize cells; similarly, E6 mutants with normal degradation activity fail to immortalize cells (Kiyono et al. 1997).
One p53-independent function of E6 is the activation of telomerase by HR E6 proteins (Klingelhutz et al. 1996). Telomerase is an enzyme with four subunits that replicates telomeric DNA at the ends of chromosomes by adding hexamer repeats. Expression of its catalytic subunit, human telomerase reverse transcriptase (hTERT), plays an essential role in regulation of telomerase activity (Liu 1999). Over successive cell divisions, the telomeres become critically shortened and dysfunctional, leading to a limited cell proliferative lifespan because of the induction of senescence and irreversible cell growth arrest. In contrast, in cancers, hTERT expression is typically reactivated resulting in reconstitution of telomerase activity (Liu 1999). Studies have revealed that E6 increases expression of endogenous hTERT levels through transcriptional activation of the hTERT promoter through the action of NFX1-123, Myc and Sp-1 (Kyo et al. 2000; Howie et al. 2009; Gewin and Galloway 2001; Katzenellenbogen et al. 2009). While this activity is not the only function necessary for efficient immortalization of the host cell, it is a crucial function of E6.
Another interaction important for the ability of E6 proteins to immortalize cells is its association with several PDZ domain–containing proteins. PDZ domains are approximately 90 amino acids in size and are binding domains for a number of proteins including post-synaptic density protein (PSD-95), Drosophila disk large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1). These domains are often found in proteins responsible for cell–cell adhesion as well as cell signaling and typically localized in areas of cell–cell contact. Studies have shown that PDZ proteins MUPP-1, hDLG, hScribble, and MAGI-1, 2, 3 bind to the extreme C-terminus of HR E6 proteins resulting in degradation of the PDZ protein (Lee et al. 1997, 2000). While PDZ protein interactions with E6 proteins may contribute to malignant progression, complete characterization of the mechanisms involved is still unclear.
HR E6 proteins have also been found to interact with various other cellular factors including paxillin, the putative calcium-binding protein E6-BP, and the interferon regulatory factor IRF-3 (Patel et al. 1999; Ronco et al. 1998). Several studies have identified many cellular binding partners for LR E6 proteins such as MCM7, Bak, zyxin, and GPS2 (Kuhne and Banks 1998; Kukimoto et al. 1998; Thomas and Banks 1999). It is clear from the characterization of numerous E6-binding partners and activities, that this viral protein is essential in the viral life cycle since knockout of E6 in genomes results in loss of ability to maintain episomes (Thomas et al. 1999).
The second oncoprotein, E7, is approximately 100 amino acids in size and is able to form dimers via its C-terminus. HR E7 proteins are comprised of three conserved regions: CR1 present at the N-terminus; CR2 containing an LXCXE motif that binds the retinoblastoma protein (Rb); and CR3, which contains two zinc finger-like motifs (Dyson et al. 1992). The CR1 and CR2 domains have sequence homology to the conserved regions CR1 and CR2 in adenovirus E1A. E7 is able to transform NIH3T3 fibroblasts by itself and with increased efficiency upon co-expression of E6. In contrast to E6, E7 oncoproteins are able to immortalize human keratinocytes when expressed alone, albeit, at a very low frequency (Howley and Lowy 2007; Munger et al. 1989; Riley et al. 2003).
A central activity of E7 proteins is their association with the Rb family of proteins (Dyson et al. 1989). Rb, p107, and p130 are the members of this family, and their expression occurs throughout the cell cycle, regulating cell cycle progression. In order to understand how E7 can circumvent Rb-regulated cell cycle progression, it is first important to appreciate how Rb regulates the cell cycle in normal circumstances. Unphosphorylated Rb proteins and the E2F/DP1 transcription factors form complexes to repress transcription of genes involved in S phase progression (DNA synthesis) or apoptosis. E2F transcription factors regulate the transcription of proteins required for normal cellular DNA synthesis. The transition from G1 to S phase is triggered when cyclin kinase complexes phosphorylate the Rb proteins causing their release from the E2F complex relieving transcriptional repression of transcription of genes involved in DNA synthesis.
E7 alters the regulation of G1/S by promoting the constitutive expression of E2F-regulated genes by binding Rb and sequestering it away from forming E2F/DP1 complexes. This relieves transcriptional repression and allows genes required for DNA synthesis to be transcribed (Edmonds and Vousden 1989; Weintraub et al. 1995). Additionally, E7 is able to target Rb for ubiquitin-mediated proteasomal degradation, which again allows for E2F-regulated genes to be constitutively transcribed, promoting DNA synthesis (Howley and Lowy 2007; Moody and Laimins 2010). Rb proteins are also responsible for controlling cell cycle exit during epithelial differentiation; hence, E7-mediated abrogation of Rb function maintains cell cycle activity. This activity is necessary for productive viral replication to occur in the differentiated epithelial cells (Thomas et al. 1999). The importance of the Rb-E7 is also essential for the virus’ ability to maintain the genome as episomes in undifferentiated cells (Longworth and Laimins 2004b).
Another way in which E7 proteins can affect cell cycle progression is via its ability to associate with cyclins and cyclin-dependent kinases (cdk) inhibitors. For example, the association of E7 with cyclins A and E as well as cdk inhibitors p21 and p27 has been characterized in various studies (Davies et al. 1993; Funk et al. 1997; Jones et al. 1997; Ruesch and Laimins 1998; Tommasino et al. 1993; Zerfass-Thome et al. 1996). These cellular proteins affect the phosphorylation status of Rb proteins, and in doing so facilitate cell cycle progression. Specifically, E7 proteins can bind cyclins A, E, p21, and p27 resulting in an increase in cyclin A and E levels while causing a decrease in p21 and p27 levels (Jones et al. 1997; Ruesch and Laimins 1998; Tommasino et al. 1993). The net effect of these interactions is to drive progression of the cell cycle, which is necessary for facilitating the HPV life cycle in differentiating epithelia.
Alteration of Rb phosphorylation status is not the only means of affecting E2F-responsive promoters as they can also be repressed by action of histone deacetylases (HDACs). Targeting of class I HDACs is another mechanism in which E7 affects cell cycle regulation (Longworth and Laimins 2004a, b; Brehm et al. 1998). HDACs are ubiquitously expressed transcriptional co-repressors that remove acetyl groups from lysine-rich N-terminal tails of histone proteins in the nucleosome. Additionally, HDACs can directly deacetylated E2F-responsive factors resulting in the loss of their function. There are three classes of HDACs, which are classified according to sequence homology and localization of the factors in the cell. Class I HDACs are localized to the nucleus and include HDACs 1, 2, 3, and 8. Class I HDACs require binding to cofactor proteins that either modify their activity or localize them to site of action. HR E7 proteins indirectly associate with class I HDACs through direct binding to the auxiliary protein, Mi2β, via sequences in the zinc finger regions at the C-terminus (Brehm et al. 1998). This HDAC/E7 interaction specifically results in increased levels of E2F-responsive transcription in differentiating cells, which allows cells to maintain cell cycle activity (Longworth and Laimins 2004b). The binding of E7 to HDACs is also important in facilitating the viral life cycle as HPV 31 genomes with mutations abrogating the E7/HDAC interaction have slower growth, are defective in maintaining genomes as episomes, and have a limited life span (Longworth and Laimins 2004b).
Many cancers exhibit increased genomic instability, and similar effects are seen in HPV-induced cancers. HR E7 expression is responsible for inducing genomic instability in these cancers. In biopsies isolated from HPV-positive cancers, high levels of aneuploidy are observed suggesting that changes in chromosome numbers may promote the progression from low-grade lesions to malignancy. During normal cell division, centrosomes coordinate equal segregation of chromosomes to the daughter cells. Expression of E7 in human keratinocytes results in an increase in the number of cells harboring abnormal centrosome numbers, indicating E7 is a major inducer of chromosome missegregation (Duensing et al. 2000). Furthermore, E7 was found to induce chromosomal abnormalities in cells deficient of p130, Rb, and p107, indicating that this action is independent of E7’s ability to bind and/or degrade Rb (Duensing and Munger 2003).
Related posts:
 Infection, Inflammation and Prevention of Cancer
Infection and Human Cancer: An Overview
of Hepatitis C Virus Infection and Liver Cancer
Oncogenic Role of Hepatitis C Virus
of Virus Infection and Human Cancer
Biology of Human Herpesvirus 8: Novel Functions and Virus–Host Interactions Implicated in Viral Pathogenesis and Replication
Infection, Inflammation and Prevention of Cancer
Infection and Human Cancer: An Overview
of Hepatitis C Virus Infection and Liver Cancer
Oncogenic Role of Hepatitis C Virus
of Virus Infection and Human Cancer
Biology of Human Herpesvirus 8: Novel Functions and Virus–Host Interactions Implicated in Viral Pathogenesis and Replication
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


