Fig. 1
The structure of hepatitis C virus genome. The HCV genome RNA encodes a polyprotein of 3,010 a.a., which is processed into structural and non-structural proteins by the cellular or viral proteases. One of the structural proteins, the core protein, shows a versatile character in experiments both in vitro and in vivo. ISDR, interferon sensitivity–determining region
The polyprotein is processed by both the cellular and viral proteases to generate the viral gene products, which are subdivided into structural and non-structural proteins. The structural proteins, which are encoded by the NH2-terminal quarter of the genome, include the core protein and the envelope proteins, E1 and E2. The E2 has an alternative form, E2-p7, though it is not clear whether or not the p7 composes the viral particle. The NS2, NS3, NS4A, NS4B, NS5A, and NS5B are the non-structural proteins that are coded in the remaining portion of the polyprotein. These include serine protease (NS3/4A), NTPase/helicase (NS3), and RNA-dependent RNA polymerase (NS5B).
The core protein of HCV occupies residues 1–191 of the precursor polyprotein and is cleaved between the core and E1 protein by host signal peptidase. The C-terminal membrane anchor of the core protein is further processed by host signal peptide peptidase (Moradpour et al. 2007). The mature core protein is estimated to consist of 177–179 amino acids and shares high homology among HCV genotypes. The HCV core protein possesses the hydrophilic N-terminal region “domain 1” (residues 1–117) followed by a hydrophobic region called “domain 2,” which is located from residue 118–170. The domain 1 is rich in basic residues and is implicated in RNA binding and homo-oligomerization. The amphipathic helices I and II spanning from residue 119–136 and residue 148–164, respectively, in domain 2 are involved in the association of HCV core protein with lipid (Boulant et al. 2006). In addition, the region spanning from residue 112–152 is associated with membranes of the endoplasmic reticulum and mitochondria (Suzuki et al. 2005). The core protein is also localized into the nucleus (Miyamoto et al. 2007; Shirakura et al. 2007) and binds to the nuclear proteasome activator PA28γ/REG γ, resulting in PA28γ-dependent degradation of the core protein (Moriishi et al. 2003). Autophagy is involved in the degradation of cellular organelles and the elimination of invasive microorganisms. Disruption of autophagy often leads to several protein deposition diseases. Recently, it has been shown that replication of HCV RNA induces autophagy in a strain-dependent manner, suggesting that HCV harnesses autophagy to circumvent cell death, and dysfunction of autophagy flux may participate in the genotype-specific pathogenesis of HCV (Taguwa et al. 2011).
3 Possible Role of HCV in Hepatocarcinogenesis
The mechanism underlying hepatocarcinogenesis in HCV infection is not fully understood yet, despite the fact that nearly 80 % of patients with HCC in Japan and 30 % of those in the world (Perz et al. 2006) are persistently infected with HCV (Kiyosawa et al. 1990; Saito et al. 1990; Yotsuyanagi et al. 2000). These lines of evidence prompted us to seek for determining the role of HCV in hepatocarcinogenesis. Inflammation induced by HCV should be considered in hepatitis viral infection: necrosis of hepatocytes due to chronic inflammation followed by regeneration enhances genetic aberrations in host cells, the accumulation of which culminates in HCC. This theory presupposes an indirect involvement of HCV in HCC via hepatic inflammation. However, this context leaves us with a serious question: Can inflammation alone result in the development of HCC in such a high incidence (90 % in 15 years) or multicentric nature in HCV infection?
The other role of HCV would have to be weighed against a rare occurrence of HCC in patients with autoimmune hepatitis in which severe inflammation in the liver persists, even after the development of cirrhosis. These backgrounds and reasonings lead to a possible activity of viral proteins for inducing neoplasia. This possibility has been evaluated by introducing genes of HCV into hepatocytes in culture with little success. One of the difficulties in using cultured cells is the carcinogenic capacity of HCV, if any, which would be weak and would take a long time to manifest itself. Actually, it takes 30–40 years for HCC to develop in individuals infected with HCV. On the basis of these viewpoints, investigation was started on carcinogenesis in chronic hepatitis C, in vivo, using transgenic mouse technology.
4 HCV Shows an In Vivo Oncogenic Activity in Mouse Studies
One of the major issues regarding the pathogenesis of HCV-associated liver lesion is whether the HCV proteins have direct effects on pathological phenotypes. Although several strategies have been used to characterize the hepatitis C viral proteins, the relationship between the protein expression and disease phenotype has not been clarified. For this purpose, several lines of mice have been established which were transgenic for the HCV cDNA (Table 1). They include the ones carrying the entire coding region of HCV genome (Lerat et al. 2002), the core region only (Machida et al. 2006; Moriya et al. 1997), the envelope region only (Koike et al. 1995; Pasquinelli et al. 1997), the core and envelope regions (Lerat et al. 2002; Naas et al. 2005), and the core to NS2 regions (Wakita et al. 1998). Although detection of mRNA from the NS regions of the HCV cDNA has been reported (Honda et al. 1999; Lerat et al. 2002), the detection of HCV NS proteins in the transgenic mouse liver has not been successful. The reason for this failure in detecting NS proteins is unclear, but the expression of the NS enzymes may be harmful to mouse development and may allow the establishment of only low-expression mice.
Table 1
Consequences of the expression of HCV proteins in mice
HCV gene | Genotype | Promoter | Protein expression | Phenotypes | References |
|---|---|---|---|---|---|
Core | 1b | HBV | Similar to patients | Steatosis, HCC, insulin resistance, oxidative stress | Shintani et al. (2004) Miyamoto et al. (2007) |
Core | 1b | EF-1a | Similar to patients | Steatosis, adenoma, HCC, oxidative stress | Machida et al. (2006) |
E1-E2 | 1b | HBV | Abundant | None in the liver | |
Core-E1-E2 | 1b | Albumin | Similar to patients | Steatosis, HCC, oxidative stress | Lerat |
Core-E1-E2 | 1a | CMV | Similar to patients | Steatosis, HCC | Naas et al. (2005) |
Structural proteins | 1b | MHC | Low in the liver | Hepatitis | Honda et al. (1999) |
Entire Polyprotein | 1b | Albumin | Only mRNA detectable | Steatosis, HCC | Lerat |
Entire Polyprotein | 1a | A1-antitrypsin | Steatosis, intrahepatic T-cell recruitment | Alonzi et al. (2004) | |
NS3/4A | 1a | MUP | None (modulation of immunity) | Frelin et al. (2006) | |
NS5A | 1a | ApoE | None (resistance to TNF) | Majumder et al. (2002) |
We have engineered transgenic mouse lines carrying the HCV genome where by introducing the genes from the cDNA of the HCV genome of genotype 1b (Moriya et al. 1997, 1998). Established are four different kinds of transgenic mouse lines, which carry the core gene, envelope genes, the entire nonstructural (NS) genes, and NS5A gene, respectively, under the same transcriptional regulatory element. Among these mouse lines, only the transgenic mice carrying the core gene developed HCC in two independent lineages (Moriya et al. 1998). The envelope gene transgenic mice do not develop HCC, despite high expression levels of both E1 and E2 proteins (Koike et al. 1995, 1997), and the transgenic mice carrying the entire NS genes or NS5A gene have developed no HCC.
The core gene transgenic mice, early in life, develop hepatic steatosis, which is one of the histologic characteristics of chronic hepatitis C, along with lymphoid follicle formation and bile duct damages. Thus, the core gene transgenic mouse model well reproduces the feature of chronic hepatitis C. Of note, any pictures of significant inflammation are not observed in the liver of this animal model. Late in life, these transgenic mice develop HCC. Notably, the development of steatosis and HCC has been reproduced by other HCV transgenic mouse lines, which harbor the entire HCV genome or structural genes including the core gene (Lerat et al. 2002; Machida et al. 2006; Naas et al. 2005). These outcomes indicate that the core protein per se of HCV has an oncogenic potential when expressed in vivo.
5 HCV Augments Oxidative Stress Production and Modulates Intracellular Signaling
There is a notable feature in the localization of the core protein in hepatocytes; while the core protein predominantly exists in the cytoplasm associated with lipid droplets, it is also present in the mitochondria and nuclei (Moriya et al. 1998). On the basis of this finding, the pathways related to these two organelles, the mitochondria and nuclei, were thoroughly investigated.
One effect of the core protein is an increased production of oxidative stress in the liver. We would like to draw particular attention to the fact that the production of oxidative stress is increased in the core gene transgenic mouse model in the absence of inflammation in the liver. The overproduction of oxidative stress results in the generation of deletions in the mitochondrial and nuclear DNA, an indicator of genetic damage (Moriya et al. 2001a).
Augmentation of oxidative stress is implicated in the pathogenesis of liver disease in HCV infection as shown by a number of clinical and basic studies (Farinati et al. 1995). Reactive oxygen species (ROS) are endogenous oxygen-containing molecules formed as normal products during aerobic metabolism. ROS can induce genetic mutations as well as chromosomal alterations and thus contribute to cancer development in multistep carcinogenesis (Fujita et al. 2008; Kato et al. 2001). Oxidative stress has been shown to be more augmented in hepatitis C than in other types of hepatitis such as hepatitis B (Farinati et al. 1995). Thus, a major role in the pathogenesis of HCV-associated liver disease has been attributed to oxidative stress augmentation, but little has been known about the mechanism of increased oxidative stress in HCV infection. Hence, it is an important issue to understand the mechanism of oxidative stress augmentation, both on generation and scavenging of ROS, which may allow us to develop new tools of therapies for chronic hepatitis C.
Other pathways in hepatocarcinogenesis would be the alteration of the expression of cellular genes and modulation of intracellular signaling pathways. For example, tumor necrosis factor (TNF)-α and interleukin-1β have been found transcriptionally activated (Tsutsumi et al. 2002). The mitogen-activated protein kinase (MAPK) cascade is also activated in the liver of the core gene transgenic mouse model. The MAPK pathway, which consists of three routes, c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK), is involved in numerous cellular events including cell proliferation. In the liver of the core gene transgenic mouse model prior to HCC development, only the JNK route is activated. In the downstream of the JNK activation, transcription factor–activating protein (AP)-1 activation is markedly enhanced (Tsutsumi et al. 2002, 2003). At far downstream, the levels of cyclin D1 and cyclin-dependent kinase (CDK) 4 are increased. Thus, the HCV core protein modulates the intracellular signaling pathways and gives advantage for cell proliferation to hepatocytes. In addition, HCV core protein suppresses the expression of suppressor of cytokine signaling (SOCS)-1, a negative regulator of cytokine signaling pathway, which may work as a tumor suppressor gene (Miyoshi et al. 2005).
Such an effect of the core protein on the MAPK pathway, combined with that on oxidative stress, may explain the extremely high incidence of HCC development in chronic hepatitis C (Fig. 2).
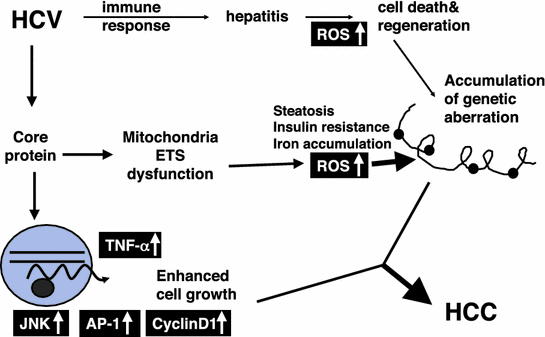
Fig. 2
Molecular pathogenesis of hepatocarcinogenesis in HCV infection. Induction of oxidative stress together with hepatic steatosis by the HCV core protein would play a pivotal role in the development of HCC. Alterations in cellular gene expressions, such as TNF-α, and those in the intracellular signaling pathways including JNK would be co-accelerators to hepatocarcinogenesis in HCV infection. ROS, reactive oxygen species; HCC, hepatocellular carcinoma; TNF-α, tumor necrosis factor-α; JNK, c-Jun N-terminal kinase; AP-1, activating protein-1; ETS, electron transfer system
6 Mitochondria as Origin of ROS Production in HCV Infection
What is the origin for the increase in oxidative stress in the liver of hepatitis C patients? The core protein is mostly localized to the endoplasmic reticulum (ER), but it is also localized to the mitochondria in cultured cells and transgenic mice (Moriya et al. 1998; Suzuki et al. 2005). In addition, the double structure of mitochondrial membranes is disrupted in hepatocytes of core gene transgenic mice. Evidence suggests that the core protein modulates some mitochondrial functions, including fatty acid β-oxidation, the impairment of which may induce lipid abnormalities and hepatic steatosis. In addition, the mitochondrion is an important source of ROS. In livers of transgenic mice harboring the core gene, increased ROS production has been observed (Moriya et al. 2001a). A recent study found, by the proteomic profiling of biopsy specimens, that impairment in key mitochondrial processes, including fatty acid oxidation and oxidative phosphorylation, and in the response to oxidative stress occurs in HCV-infected human liver with advanced fibrosis (Diamond et al. 2007). Therefore, it is probable that the HCV core protein affects mitochondrial functions, since such pathogenesis is observed in HCV core transgenic mice, cultured cells expressing the core protein (Korenaga et al. 2005), and HCV-infected patients.
The recent progress in proteomics has opened new avenues for disease-related biomarker discovery. We performed a two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) of mitochondria isolated from HepG2 cells stably expressing the HCV core protein and identified several proteins of different expressions when compared with control HepG2 cells. Among upregulated proteins in the core-expressing cells, we focused on prohibitin, which functions as a mitochondrial protein chaperone, and found that the core protein interacts with prohibitin and represses the interaction between prohibitin and subunit proteins of cytochrome C oxidase (COX), which leads to a decrease in the expression level of the proteins and in COX activity.
Prohibitin, a mitochondrial protein chaperone, was identified as an upregulated protein in core-expressing cells. Prohibitin is a ubiquitously expressed and highly conserved protein that was originally determined to play a predominant role in inhibiting cell cycle progression and cellular proliferation by attenuating DNA synthesis (Mishra et al. 2005). It exists in the nucleus and interacts with transcription factors that are vital in cell cycle progression. In core-expressing cells, prohibitin was also detected in the nucleus, and its expression level was also higher than that in control cells. Mitochondrial prohibitin acts as a protein chaperone by stabilizing newly synthesized mitochondrial translation products through direct interaction (Nijtmans et al. 2000). We examined the interaction between prohibitin and mitochondria-encoded subunit II of COX and found a suppressed interaction between these proteins in core-expressing cells. In addition, there are several studies that showed the association of prohibitin with the assembly of mitochondrial respiratory complex I as well as complex IV (COX) (Nijtmans et al. 2000). Complex I also consists of both nuclear and mitochondrial DNA-encoded subunits; therefore, it is probable that the assembly and function of complex I are impaired by the core protein. With respect to the complex I function, we previously found a decreased complex I activity in core-expressing cells. Other groups have also shown that complex I activity is decreased in cultured cells (Piccoli et al. 2007). From these findings, the interaction between prohibitin and the core protein may impair the function of complex I as well as complex IV, leading to an increase in ROS production. In fact, the suppression of the prohibitin function is shown to result in an increased production of ROS (Theiss et al. 2007), a phenomenon observed in core-expressing cells used in this study as well as in the liver of core gene transgenic mice (Moriya et al. 2001a). Very interestingly, the liver-specific deletion of prohibitin resulted in the morphological abnormality and HCC (Ko et al. 2010).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


