Fig. 7.1
IL-12 links innate and adaptive antitumor immunity. IL-12 utilizes several mechanisms to induce antitumor effects. IL-27 activates innate effectors, such as NK cells, NKT cells, and γδ − T cells and promotes their cytolytic activity and cytokine production. IL-12 induces IFN-γ production in macrophages that can have a cytotoxic effect on tumor cells. IL-12 induces the production of antiangiogenic molecules from endothelial cells. In addition, IL-12 has a direct toxic effect on the some tumor cells. Furthermore, IL-12 secretion by DCs can induce adaptive arms of antitumor immunity. IL-12 can augment Th1 response necessary for cellular immune response. Il-12 stimulates the differentiation and lytic capacity of CTL and promotes immune memory. Finally, IL-12 can mediate antibody-mediated tumor clearance via B-cell activation
In addition to its effect on NK cell cytotoxicity, IL-12 enhances T-cell-mediated cytotoxicity and has an enhancing effect on CD8+ T cells [23]. DCs play a crucial role in facilitating the interaction between CD4+ T cells and antigen-specific CD8+ T cells. Priming of CTL is enabled by the ligation of CD40 on DC and its ligand CD154 on activated CD4+ T cells [24, 25]. The induction of IL-12 synthesis that occurs as a result of CD40 ligation suggests an important role for IL-12 in the molecular mechanisms responsible for CTL priming [26]. It was then shown that IL-12, in the presence of antigen, acts directly on naive CD8+ T cells to promote clonal expansion and differentiation [27]. In addition, priming of CD8+ T cells in the absence of IL-12 rendered them unresponsive to the same antigen [28]. Agonistic CD40 antibodies (Abs) were shown to substitute the function of CD4+ T cells in murine models of T-cell-mediated immunity, resulting in rapid expansion of CTLs that cleared established lymphomas and provided long-term protection against tumor rechallenge [29, 30]. These observations provided an explanation for the impaired tumor antigen-specific CTL activation in CD40-deficient mice and confirmed the key role of the CD40-IL-12 pathway in the regulation of antitumor immunity. A series of experiments, conducted by different groups, indicated that the injection of IL-12 directly into subcutaneous tumors results in CTL response against the tumor in mice [31–33].
The rejection of tumors requires CD8+ T cells whose activation and maintenance depends on CD4+ T cells. Upon stimulation, naïve CD4+ T cells differentiate into different lineages of T helper subsets including Th1, Th2, Th17, and Tregs. These distinct CD4+ T-cell subsets have varied impact on tumor growth. While Th1 cells promote CD8+ T-cell-mediated immunity to tumors, the other CD4+ T-cell subsets Th2 and Tregs negatively regulate CD8+ T-cell function. In the presence of IL-12, naïve CD4+ T cells differentiate into IFN-γ-secreting Th1 cells [34]. Th1 cytokines, IL-2, and IFN-γ, stimulate the cytolytic activity of NK cells. High production of IFN-γ by CD8+ T cells and a Th2 to Th1 shift in the cytokine secretion profile of CD4+ T cells were also seen in the IL-12-treated mice [35]. By altering the balance between Th1 and Th2 cytokines, IL-12 plays a critically important role in antitumor immune responses. A shift from Th1 to Th2 cytokine production has been reported in progressive cancer patients, and a vaccine inducing Th2 to Th1 shift in a murine model of tumor was shown to induce tumor rejection [36]. In addition, Th2 cytokines have been shown to accelerate tumor growth in multiple experimental models [37]. In fact, CD4+ T cells can directly interact with CD8+ T cells via CD40-CD154 interactions [38], which directly contrast with the early notion that CD4+ and CD8+ T cells are brought together on the same antigen-presenting cell for the effective delivery of IL-2 to neighboring CD8+ T cells. Moreover, a full CD8+ T-cell response is elicited by a temporal release of IL-2 from CD4+ T cells, which is consistent with the findings that neutralization of IL-2 significantly limits CD8+ T-cell growth [39]. IL-12 also plays an important role in the establishment of memory CD8+ T cells [40]. A strong specific CTL response was observed in patients with advanced melanoma after administration of IL-12. The number of tumor-specific CTL increased in the circulation, and influx of specific memory CD8+ T cells into metastasized lesions was documented [41]. Additionally, IL-12 was shown to stimulate humoral immunity. In a model of colon carcinoma, vaccination with IL-12-transduced tumor cells cured 40 % of tumor-bearing mice. Favorable antitumor responses were related to the synthesis of Abs against tumor antigens inducing tumor cell lysis in a complement-dependent cytotoxicity assay [42].
The ability of IL-12 to facilitate cell-mediated immune responses, including enhancement of NK cytotoxicity, generation of CTL, and DC activation, suggests its role in both the innate and adaptive immunity resistance mechanisms against tumors. Experimental studies of systemic administration of the cytokine have indicated that IL-12 exerts potent antitumor activity against a variety of metastatic tumors and can even prevent spontaneous tumor development in HER-2/neu transgenic mice. In addition, models based on intra-tumor cytokine delivery or in vivo transfer of cytokine-secreting tumors have indicated that IL-12 has significant dose-dependent antitumor activity against a wide spectrum of murine tumors including melanoma and breast, ovarian, and bladder tumors [17, 43, 44]. All these studies have demonstrated that IL-12 can inhibit tumor growth and improve the survival of tumor-bearing animals that are dependent on not only its ability to activate the innate and adaptive arms of antitumor immunity but also through its antiangiogenic activity.
7.2.1.3 IL-12 and Angiogenesis Inhibition
Accumulating evidence indicates that the antitumor effects of IL-12 are mediated, at least in part, through mechanisms involving angiogenesis and its direct effects on tumors. Angiogenesis is an essential process for tumor growth and metastases. In addition, it is the result of a complex balance between angiogenic and antiangiogenic factors. The balance between angiogenic and angiostatic molecules in the tumor microenvironment can determine tumor growth and survival. The antiangiogenic properties of IL-12 were first observed by Voest et al. who demonstrated that IL-12 treatment almost completely inhibited neovascularization in immunocompetent mice, severe combined immunodeficient mice, and T-cell-deficient nude mice [45]. However, suppression of angiogenesis by IL-12 was dependent on its ability to induce IFN-γ expression. Accordingly, administration of IFN-γ reproduced the antiangiogenic effects promoted by IL-12. Moreover, it was shown that inhibition of tumor growth by IL-12 or IFN-γ required an intact signaling from IFN-γ receptors expressed in neoplastic cells. This indicated that IL-12 could inhibit tumor growth by inducing neoplastic cells to produce antiangiogenic factors. Two of the most relevant factors were identified as the IFN-γ-inducible genes, IFN-inducible protein 10 (IP-10) and monokine induced by interferon-γ (MIG) [46, 47]. Local and systemic treatment with IL-12 was associated with the expression of IFN-γ, IP-10, and MIG in the tumor; in addition, intra-tumor delivery of MIG into subcutaneously growing tumor in nude mice led to tumor necrosis associated with vascular damage. Administration of neutralizing Abs to IP-10 and MIG substantially reduced the antitumor effects of IL-12 [48]. IP-10 and MIG interact with their receptor CXCR3 to mediate their angiostatic activity. These results support the notion that these chemokines, both ligands of the receptor CXCR3, contribute to the antitumor effects of IL-12 through their inhibitory effect on tumor vasculature. In addition to IFN-γ stimulation, IL-12 promotes the expression of interferon regulatory factors 1 (IRF-1) and 4 (IRF-4), which are necessary for Th1 cell differentiation [49]. IRF-1 has tumor suppressor activities in cancer cells in vitro and decreases the tumorigenicity of cells inoculated into athymic nude mice [50, 51]. Similarly, IRF-4 suppresses c-Myc-induced leukemia in animal models and inhibits BCR/ABL-induced B-cell acute lymphoblastic leukemia [52, 53].
Emerging evidence indicates the involvement of lymphocyte-endothelial cell crosstalk at the beginning of the process of angiogenesis inhibition by IL-12. It has been shown that neutralization of NK cell function reversed IL-12 inhibition of angiogenesis in athymic nude mice. Immunohistochemistry analysis revealed that neovascularization inhibited by IL-12 displayed accumulation of NK cells and IP-10-positive cells. In addition, experimental Burkitt lymphomas treated locally with IL-12 displayed tumor tissue necrosis, vascular damage, and NK cell infiltration surrounding small vessels [54]. These results documented that NK cell cytotoxicity of endothelial cells is a potential mechanism by which IL-12 can suppress neovascularization. The antiangiogenic program activated in lymphocytes by IL-12 can also directly affect gene expression in neoplastic cells. In fact, upregulation of signal transducers and activators of transcription-1 (STAT-1) and angiopoietin 2 together with down-modulation of vascular endothelial growth factor (VEGF) has been observed in neoplastic cells exposed to soluble factors released by IL-12-stimulated lymphocytes [55]. In addition, IL-12 treatment reduced the production of metalloproteases, playing a role in matrix remodeling, a process required during neoangiogenesis [56]. Moreover, the activation of integrin αVβ3 on endothelial cells is reduced by the IL-2-induced IFN-γ, which leads to decreased endothelial cell adhesion and survival [57]. IL-12-induced secretion of IFN-γ leads to an increase in p53 activity, which subsequently results in tumor suppression due to the induction of apoptosis in cancer cells [58]. Furthermore, IL-12 dramatically decreased tumor-supportive activities of tumor-associated macrophages (TAMs), which are involved in tumor angiogenesis and metastasis. The antiangiogenic mechanisms mediated by IL-12 are complex and dependent not only on the direct effect on endothelial cells of the proinflammatory cytokine/chemokines induced by IL-12 but also on the recruitment of immune effector cells such as NK and T cells.
7.2.1.4 Regulation of IL-12 in Tumor Microenvironment
Although controlled Th1 and CTL responses can exert a significant antitumor immunity, the same responses, if exaggerated, may result in host-tissue destruction and autoimmunity. Therefore, as a part of immune homeostasis, the inflammatory responses need to be counter-regulated. Tregs play a major role in controlling unwanted immune response to self-antigens [59]. Studies have revealed a significant role for Tregs in defective immune responses to tumor antigens. Treg functions are mediated in part through secretion of immunosuppressive cytokines IL-10 and TGF-β. Both TGF-β and IL-10 can inhibit DC antigen presentation, IL-12 secretion, and effector functions of both CD4+ and CD8+ T cells [12]. Thus, it is possible that as an immunosuppressive environment develops in the growing tumor, DCs secreting IL-12 become scarce. This might be due to an absence of DC activation signals, CD40, or inhibition of activated CD4+ T cells which could themselves activate DC. Moreover, the CD40-CD40L interaction between DCs and T cells leads to the induction of not only IL-12 but also IL-10, a pro-tumor cytokine that may act in an autocrine or a paracrine manner to downregulate IL-12 secretion from DCs [60]. Indeed, reduced CD40 expression on DC or CD40-L on T cells from tumor-bearing hosts may explain the reason for reduced levels of IL-12 observed in patients with cancer [61]. In accordance with this, reduced expression of IL-12 was observed in patients with advanced cancer types including glioblastoma, renal cell carcinoma, head and neck squamous cell carcinoma, gastric cancer, melanoma, colorectal cancer, hepatocellular carcinoma, and gastric cancer [15]. Moreover, IL-12 production by stimulated peripheral blood mononuclear cells decreased significantly in patients with gastric and colorectal cancer with advanced disease. In addition to the immunosuppressive cytokines TGF-β and IL-10, other factors present in the tumor microenvironment can downregulate IL-12 production, for example, prostaglandin E2 (PGE2) produced by tumor cells or tumor-associated host cells (macrophages, endothelial cells, and stromal cells) known to inhibit IL-12 production [62].
7.2.1.5 Clinical Studies with IL-12
Based on the provocative preclinical studies, IL-12 was evaluated in patients with different malignancies. However, clinical experience with IL-12 in humans is limited. Several phase I clinical trials of IL-12 in patients with solid tumors and hematological malignancies have been reported [63]. IL-12 administration in patients with advanced colorectal cancer (CRC), melanoma, and renal cell carcinoma resulted in only one partial response (renal cell carcinoma) and one transient complete response (melanoma), among the 40 enrolled patients. However, common signs and symptoms of toxicity such as fever/chills, nausea, vomiting, fatigue, and headache were observed [64]. Administration of IL-12 resulted in stabilization of the disease in several renal cancer patients and partial regression of a metastatic lesion, but has not proceeded further in clinical development due to signs and symptoms of toxicity, including fever, vomiting, and elevation of hepatic enzymes [65]. Clinical trials of IL-12 treatment in combination with rituximab in patients with B-cell non-Hodgkin lymphoma (NHL) did not result in clinical response [66]. However, several clinical studies revealed positive results with IL-12 administration. During IL-12 treatment in patients with NHL, 21 % of the patients had a partial or complete response without major side effects [67]. Similarly, subcutaneous IL-12 treatment resulted in complete response in 56 % of the treated patients with T-cell lymphoma with minor toxicity [68]. Furthermore, clinical trials on metastatic melanoma revealed that IL-12 administration induces tumor shrinkage in patients accompanied with increased frequency of circulating antitumor CTLs [41]. The low efficacy of IL-12 in the abovementioned clinical trials may be due to an immunosuppressive microenvironment in advanced tumors. In addition, IL-12 may self-limit its own therapeutic efficacy by inducing IL-10 and other suppressive factors. For example, IFN-γ induced by IL-12 can activate immunoregulatory molecules such as programmed death ligand-1 (PD-L1) and indoleamine 2,3 dioxygenase (IDO) on a variety of cells (DC, T cells, and endothelial cells) [69]. Both PD-L1 and IDO can abrogate antitumor immunity through various mechanisms. Furthermore, other factors such as environment and diet may alter the effectiveness of IL-12-mediated anticancer immunity. Although systemic administration of IL-12 in patients is limited by its significant toxicity, emerging studies in animal models indicate that IL-12 in combination with other cytokines boosts antitumor immunity without any toxic side effects [44]. Thus, selective targeted delivery of IL-12 to tumors and/or reducing the dose of IL-12 while combining it with other therapeutics may yield better outcome.
7.2.2 IL-27
7.2.2.1 Overview
IL-27 is a member of the IL-12 family cytokine that plays potent antitumor effects against various tumor models via different mechanisms, depending on the characteristics of each tumor [70]. Unlike IL-12, IL-27-mediated antitumor functions are independent of IFN-γ, and IL-27-treated mice do not manifest any toxic side effects. IL-27 is mainly produced by activated APCs including DCs and macrophages. DCs secrete IL-27 on exposure to physiological stimuli such as type I and type II interferons (INF) and CD40 [71–73]. In addition, IL-27 expression is induced in APCs upon stimulation by various TLR ligands such as poly(I:C), lipopolysaccharide (LPS), and CpG-DNA, which are agonists of toll-like receptors (TLR)3, TLR4, and TLR9, respectively [74–76].
7.2.2.2 IL-27 in Antitumor Immunity
IL-27 has a wide array of functions necessary for the induction of antitumor immune response. IL-27 has been shown to act on NK cells to enhance their cytotoxic activity both in vitro and in vivo; in addition, therapeutic administration of IL-27 increased NK cell susceptibility of tumors [77]. By activating NK cells, IL-27 might enhance adaptive immunity to tumors in part; the killing of tumor target by NK cells could provide DCs with increased access to tumor antigens; thus, IL-27 serves as a link between innate and adaptive antitumor immunity. In addition to NK cell activation, IL-27 acts on CD8+ T cells and induces the generation of CTL through enhancing the expression of effector molecules such as granzyme B and perforin [78]. Similar to mice, IL-27 promotes IFN-γ and granzyme B production from human CD8+ T cells [79]. The overexpression of IL-27 in highly immunogenic murine tumor cells facilitated CTL development with enhanced IFN-γ production [80, 81]. In line with these observations, IL-27R−/− mice failed to regulate tumor growth in vivo, reiterating the importance of IL-27 signaling in the generation of antitumor immunity [82]. Most recently, DC-derived IL-27 has been shown to induce NK and NKT cell-dependent antitumor immunity against methylcholanthrene-induced fibrosarcoma and transplanted B16 melanoma [83]. Moreover, IL-27 in combination with other cytokines such as IL-2 and IL-12 boosts antitumor immunity by contributing to the development of CTLs and NK cells [84].
In addition to the direct effect of IL-27 on CD8+ T-cell activation, the influence of IL-27 on CD4+ T-cell responses might provide further therapeutic opportunities. Initial studies have indicated that IL-27 leads to the differentiation of Th1 cells [85]. IL-27 synergizes with IL-12 to enhance IFN-γ production [86]. Moreover, it has been shown that IL-27 inhibits Th2 polarization of naïve CD4+ T cells and suppresses Th2 cytokine production from in vitro polarized Th2 cells [87–89]. By altering the balance between Th1 and Th2 cytokines, IL-27 plays a critically important role in antitumor immune responses. In line with this, a recent study confirmed IL-27’s capability in the reversion of the Th2 polarization of in vivo primed lymphocytes from pancreatic cancer patients [90]. IL-27-dependent enhancement of preexisting antigen-specific Th1 responses has also been demonstrated [36]. IL-27 may promote tumor regression through the inhibition of Tregs. IL-27 inhibits the generation of Foxp3+ Tregs both in vitro and in vivo. IL-27 blocks Treg differentiation through a mechanism that is at least partially dependent on STAT-3 [91, 92]. In addition, IL-27 can limit Treg cell expansion by inhibiting IL-2, a cytokine necessary for Treg development [93]. In a murine model of neuroblastoma, IL-27 has been shown to inhibit IL-2-induced Treg expansion in the tumor, promoting antitumor immune responses [84]. IL-27 also induces tumor-specific Ab response which cooperatively elicits ADCC activity [94].
7.2.2.3 Direct Effect of IL-27 on Tumors
IL-27 possesses multiple antitumor effects mediated by mechanisms involving angiogenesis and its direct effects on tumors. IL-27 has been shown to have antiproliferative activities which inhibit tumor growth and metastasis in murine melanoma [95]. The major antitumor role of IL-27 relies on its antiangiogenic property of surrounding endothelial cells and fibroblasts. IL-27 significantly inhibited tumor growth in SCID mice through the induction of antiangiogenic factors such as IP-10 and MIG from endothelial cells [96]. Consistent with these results, IL-27 has been shown to directly act on human umbilical cord endothelial cells and induce production of the antiangiogenic chemokines such as IP-10 and MIG [97]. IL-27 strongly inhibited tumor growth of primary multiple myeloma (MM) cells through inhibition of angiogenesis [98]. In addition, IL-27 downregulated a wide panel of proangiogenic genes, including matrix metalloproteinase-9 (MMP-9), TGF-β, and VEGF with a concomitant upregulation of the angiostatic chemokines IP-10 and MIG.
IL-27 may further promote tumor regression through the inhibition of a proangiogenic cytokine IL-17. IL-27 suppresses the Th17 key transcription factor RORγt and thus inhibits expression of IL-17 by T cells both in humans and mice [99, 100]. Accordingly, mice deficient in either the IL-27 EBI3 subunit or IL-27R have increased levels of IL-17 [101]. Among the Th17 suppressive molecules found in the tumor microenvironment, IL-27 is one of the most potent inhibitors of Th17 differentiation. IL-27 can be induced in tumor-infiltrating DCs by galactin-1, IFN-γ, and apoptotic tumor cells in the tumor microenvironment [71, 102, 103]. However, the proangiogenic molecules which dominate the microenvironment in advanced tumors can limit the availability of IL-27. Osteopontin (OPN), a proinflammatory cytokine, inhibits the expression of IL-27 in DCs while inducing Th17 differentiation [72]. OPN promotes tumor growth through mechanisms involving angiogenesis, tumor migration, and metastasis, suggesting that OPN may release the brake on Th17 cell responses by suppressing IL-27 in DCs. Both OPN and IL-27 are expressed in DCs and macrophages; thus, the outcome of Th17 accumulation in tumor microenvironment may depend on the fine balance with other myeloid cell populations especially tumor-infiltrating macrophages and DCs expressing OPN and IL-27.
7.2.2.4 Advantages of IL-27 Over IL-12 in Tumor Immunity
IL-27-mediated antitumor mechanisms are complex. Similar to IL-12, IL-27 utilizes effector mechanisms of innate and adaptive immunity to mediate antitumor immunity. IL-27 promotes tumor immunity through the induction of Th1 and CTL responses while inhibiting immunosuppressive Th2 and Tregs. Unlike IL-12, IL-27-mediated antiangiogenic functions are independent of IFN-γ. Thus, IL-27-treated mice are not observed with any toxic side effects [104]. The central role of IL-27 in orchestrating both the innate and adaptive arms of immunity together with multiple antiangiogenic functions explains the essential contribution of this molecule to the development of antitumor immunity against both high and poor immunogenic tumors. These observations together with the lack of toxicity observed in vivo in preclinical trials with IL-27 treatment highlight the enormous therapeutic potential of this approach.
7.3 Cytokines in Immune Tolerance to Cancer
Although certain cytokines produced in the tumor microenvironment can function to inhibit tumor growth, others can promote tumor growth and progression. Several cytokines were found to serve as growth and survival factors that act on premalignant cells, stimulate angiogenesis and metastasis, and maintain tumor-promoting immunosuppression and inflammation.
7.3.1 TGF-β
7.3.1.1 Overview
Transforming growth factor-β (TGF-β) is a pleiotropic cytokine with broad tissue distribution that plays critical roles during embryonic development, normal tissue homeostasis, and cancer [105]. Elevated TGF-β serum concentrations were observed in patients with different malignancies and were associated with poor prognosis. TGF-β is released not only by a variety of cells in human and murine tumors including T cells, macrophages, and DCs but also by tumor cells themselves [106]. Almost all human cell types are responsive to TGF-β, which signals through type I and type II TGF-β receptors (TβRI and TβRII, respectively). Upon binding of TGF-β to TβRII, TβRI is recruited and activated to phosphorylate the downstream mediators, SMAD2 and SMAD3. Phosphorylated SMAD2 and SMAD3 combine with SMAD4 to enter the nucleus to modulate gene transcription [107].
The function of TGF-β in cancer is complex. TGF-β can act as a tumor suppressor or a tumor promoter depending on the stages of tumor development. Initially, it acts as a tumor suppressor since it induces apoptosis and inhibits the growth of normal and premalignant tumor cells [108]. At later stages of tumor progression, TGF-β acts as a tumor promoter. At this stage, cancer cells protect themselves and tend to acquire resistance to TGF-β growth inhibitory signals which is an important reason for the shift from being a tumor suppressor to a tumor promoter. Subsequently, cancer cells start secreting nonphysiological levels of TGF-β in an autocrine and paracrine manner which may affect the differentiation of the tumor cells and the surrounding cellular environment, respectively, leading to development of the tumor and metastasis [108]. Notably, TGF-β induces epithelial-mesenchymal transition (EMT), whereby epithelial tumor cells acquire an invasive, mesenchymal-like phenotype accompanied by changes in the expression of cell-cell adhesion molecules and secretion of metalloproteinases, leading to metastasis [109, 110]. The potent regulatory activity of TGF-β on immune cell functions represents an important mechanism of immune tolerance to tumors. The presence of TGF-β in the microenvironment of the developing tumor is predicted to disable effective immunosurveillance by multiple mechanisms, most of which converge on the impairment of tumor cell killing by innate and adaptive immune cells (Fig. 7.2).
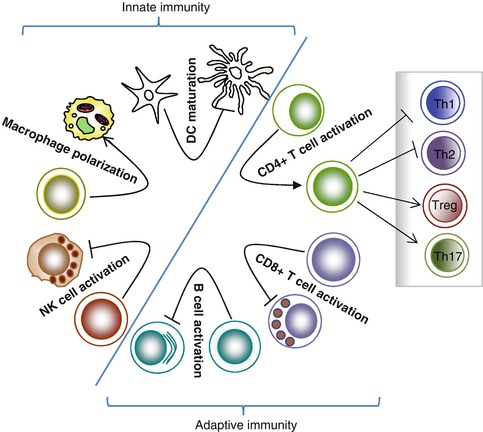
Fig. 7.2
TGF-β-mediated immunosuppression. TGF-β affects components of both innate and adaptive immune systems. TGF-β inhibits NK cell activation and its effector function. In addition, TGF-β inhibits DC maturation and antigen-presenting function while promoting polarization of M2 macrophages. TGF-β inhibits CD8+ T-cell-mediated antitumor immune response. TGF-β also has a significant impact on CD4+ T-cell differentiation and function. TGF-β induces Treg and Th17 differentiation while inhibiting Th1 and Th2 differentiation. Furthermore, TGF-β inhibits B-cell proliferation and antibody secretion
7.3.1.2 Effect of TGF-β on Innate Immunity to Tumors
TGF-β is an important regulator of NK cell function, being a potent antagonist of IL-12-induced production of IFN-γ in NK cells [111]. In addition, TGF-β inhibits the activity of NK cells by limiting expression of the activating receptor NKG2D, NKp30, and DNAM-1 [112]. In fact, reduced expression of NKG2D is associated with elevated levels of TGF-β in patients with cancer. It has been shown that surface-bound TGF-β on MDSCs can inhibit NK cell cytolytic activity against mammary adenocarcinoma [113, 114]. Moreover, TGF-β has been shown to suppress MHC class I and MHC class II expression in a number of cell populations [115–117]. Importantly, the TGF-β-dependent decrease of MHC class I expression in tumor cells has been shown to result in reduced tumor cell lysis by NK cells [117]. Although NK cells are the major innate effectors, they require activation by DCs. TGF-β has been demonstrated to impair DC function both in vitro and in vivo. TGF-β inhibits upregulation of critical co-stimulatory molecules on the surface of DCs and reduces cytokine production and their antigen-presenting capacity [118, 119]. TGF-β can immobilize DCs, thereby interfering with their migration and the transport of antigen to draining lymph nodes for presentation to T cells. Moreover, TGF-β can also induce apoptosis of DCs [120]. In recent years, more correlative clinical data has supported the inhibitory role of TGF-β in the observed defects in cancer. Increased serum TGF-β in human colorectal cancer correlates with reduced circulating DCs [121]. Moreover, tumor-infiltrating DCs both secrete and respond to TGF-β, in either an autocrine or paracrine manner. These TGF-β-secreting DCs promote the formation of Tregs [122] that potently inhibit the function of other T cells, and that Treg production of TGF-β can inhibit NKG2D-mediated NK cell cytotoxicity, thereby enhancing tumor growth and metastasis. In addition to DCs, TGF-β can suppress or alter the activation and function of other innate immune cells such as NKT cells, macrophages, and neutrophils [106].
Macrophages, the predominant leukocyte, play a key role in tumor growth. The role of tumor-associated macrophages (TAMs) in tumors is controversial [123]. TAMs originate from recruited myeloid cells, such as blood monocytes or MDSCs by a number of chemoattractants produced by tumor cells and stromal cells. Tumor-derived chemokine CCL2 is critical for the recruitment of macrophages to the tumor site [124]. Macrophages can exert different properties when polarized with distinct inducers. Differential cytokine production is a key feature of polarized macrophages. When stimulated with IFN-γ, M1 macrophages secrete high levels of IL-12, but low levels of IL-10. In contrast, M2 macrophages express high levels of IL-10 but low levels of IL-12 [125]. Due to their different cytokine profiles, these polarized macrophages have distinct functions. For example, the IL-12 produced by M1 macrophages can promote the differentiation of Th1 cells, which can improve antigen phagocytosis and contribute to antitumor immunity. Whereas, the IL-10 expressed by M2 macrophages can promote the production of IL-4 and IL-13 by Th2 cells. Both IL-4 and IL-13 have been shown to impair antitumor T-cell responses. TGF-β promotes tumor-associated macrophage polarization to an M2 vs. M1 phenotype, which further promotes TGF-β production and deepens immunosuppression [126]. In most tumors, the infiltrated macrophages are considered to be of the M2 phenotype. TAMs orchestrate various aspects of cancer, such as tumor progression, angiogenesis, metastasis, and immunosuppression [127]. It has been shown that NKT cells can suppress CTL responses through mechanisms involving TGF-β. Therefore, blockade of TGF-β signaling not only enhances the frequency of antitumor CTLs but also restores the activities of the cytolytic machinery and prevents NKT cell-mediated immunosuppression [128]. Furthermore, TGF-β also inhibits effector functions of other innate immune cells such as γδ-T cells [106]. Thus a dampened innate immune response leads to poor adaptive immunity, resulting in persistence of the tumor.
7.3.1.3 Effect of TGF-β on Adaptive Immunity to Tumors
The presence of TGF-β in the tumor microenvironment can have a profound impact upon antitumor activity of T cells. It has been shown that TGF-β can suppress CTL differentiation and CTL-mediated lysis of tumor cells [129, 130]. TGF-β acts on CTLs to specifically repress the expression of different cytolytic effector molecules such as perforin, granzyme A, granzyme B, Fas ligand (FasL), and IFN-β, which are collectively responsible for CTL-mediated tumor killing [131]. Blockade of TGF-β in tumor models has been shown to reduce tumor burden by improving CD8+ T-cell-mediated tumor immunity [131]. Furthermore, TGF-β could suppress IL-2 production and IL-2-induced T-cell differentiation [132]. Tumor cells transfected with TGF-β were shown to attenuate the efficacy of DC-based tumor vaccines [118]. In addition, TGF-β functionally regulates the differentiation of T helper cell subpopulations both in vitro and in vivo. TGF-β inhibits Th1 and Th2 cells, whereas it promotes Treg and Th17 cell differentiation [133]. Most recently, TGF-β has also been shown to play an important role in the development of IL-9-secreting Th9 cells [134].
Although there are many sources of TGF-β in the tumor microenvironment, it has been shown that Tregs can provide a significant source of TGF-β responsible for attenuation of tumor antigen expanded CTLs. Tregs hamper the functions of Th1, CD8+ T cells, NK cells, DCs, and other key effector cells of antitumor immunity [106]. Accordingly, Treg-mediated immunosuppression has been proposed to be one of the important mechanisms involved in tumor immune evasion. An accumulation of Tregs in tumors can dampen T-cell immunity to tumors and is thus the main obstacle to successful immunotherapy [59]. The frequency of Tregs present in the peripheral blood of patients with various cancers is higher than that of normal population [135]. Notably, Tregs isolated from peripheral blood and solid tumors remain suppressive to T-cell activation in vitro [136]. Likewise, Tregs from tumor-bearing mice inhibited tumor rejection, indicating that Treg cells suppress tumor-specific immunity and limit antitumor resistance. In contrast, depletion of Tregs with anti-CD25 Ab in animal models enhances antitumor immunity and tumor regression, further suggesting the involvement of Tregs in tumor growth. Furthermore, when tumor-specific CD8+ T cells were adoptively transferred with either Tregs or CD4+CD25− T cells into hosts with tumor, CD8+ T-cell-mediated immunity was abolished in those receiving Tregs but not CD4+CD25− T cells [137, 138]. Collectively, these studies provide strong evidence that Tregs can attenuate the antitumor immunity by downregulating the antitumor immune responses and ultimately facilitating the development of cancer.
7.3.1.4 TGF-β, Treg, and Tumor Angiogenesis
Angiogenesis and tumor-associated immunosuppression are hallmarks of tumorigenesis. The association between angiogenesis and immunosuppression is related to hypoxia which induces both angiogenesis and immunosuppression via activation of hypoxia-induced factor 1 (HIF-1). The induction of VEGF during hypoxia is primarily mediated by HIF-1. HIF-1-induced VEGF promotes angiogenesis by inducing the recruitment of various proangiogenic bone marrow-derived cells including endothelial progenitors and myeloid cells [139]. Although hypoxia-VEGF axis has been thought to be solely involved in vascular growth and permeability, recent studies suggest its role in immunosuppression in the tumor microenvironment. Within the tumor microenvironment multiple cell types with established roles in immunosuppression have been shown to promote angiogenesis. Among the immunosuppressive cell types found in the tumor microenvironment, Tregs are considered pivotal regulators of immunosuppression. Tregs can traffic to tumors from the periphery under the influence of chemokines in the tumor microenvironment. It has been shown that tumor hypoxia leads to the recruitment of Tregs via chemokine CCL28 [140]. Forced expression of CCL28 in mouse tumor cells resulted in accelerated tumor growth and Treg accumulation associated with increased VEGF levels and angiogenesis. In addition, Tregs were shown to express CCR4, the receptor for CCL22, and can therefore migrate to CCL22 present in the tumor microenvironment [141, 142]. Beyond recruitment of Tregs through chemokines, the tumor microenvironment promotes the continued expansion of Tregs and the generation of Tregs due to a tumor microenvironment rich in TGF-β. The recruited Tregs in turn dampen the antitumor immune response and promote angiogenesis. The accumulation of Tregs at tumors has been correlated with VEGF overexpression and increased angiogenesis in cancers, providing evidence for an association between Tregs and angiogenesis [143, 144]. Tregs can contribute to tumor angiogenesis through different mechanisms. They promote angiogenesis indirectly by suppressing Th1 cells that release angiostatic cytokine IFN-γ, as well as interferon-induced chemokines such as CXCL9 and CXCL10. Indeed, Tregs have been shown to promote tumor angiogenesis by specifically inhibiting tumor-reactive T cells. Tregs can significantly contribute to the direct promotion of tumor angiogenesis through the induction of VEGF and endothelial cell proliferation [144]. Additional therapeutic opportunities may be provided by Tregs’ capability in suppressing tumor-specific immunity while promoting tumor angiogenesis by well-planned manipulations of Tregs, including depletion, blocking trafficking into tumors, and reducing their differentiation and suppressive mechanisms. It will be beneficial to tumor eradication by combining this strategy with various current therapeutic approaches. In an early phase I clinical trial in patients with metastatic breast cancer, the anti-CD25 Ab daclizumab significantly depleted Tregs and enhanced the immunogenicity of a cancer vaccine [145]. In addition, blocking Treg function using Abs targeted against glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) is under clinical evaluation in patients with cancer.
7.3.1.5 TGF-β in Clinical Trials
As a result of the wide variety of effects of TGF-β on tumorigenesis, blockade of TGF-β and its signaling pathway can be a potent approach to improve tumor immunity. There are many TGF-β signaling antagonist agents under development at both the preclinical and clinical stages. Mice with fully or partially disrupted TGF-β function have phenotypes with severe self-reactive immune responses [146, 147]. However, clinical trials of TGF-β antagonists, such as a monoclonal Ab or small molecules that interfere with TGF-β receptor signaling, in cancer patients have been tested and are ongoing. In phase I/II clinical trials, intra-tumoral administration of AP-12009, an antisense oligonucleotide to TGF-β, resulted in a significant increase of survival time [148]. Some clinical benefit without apparent induction of autoimmune disease was found in a clinical trial of a human monoclonal anti-TGF-β in melanoma patients [149, 150]. In addition, a vaccine that contains allogeneic tumor cells that are modified to express antisense TGF-β has been tested in a phase I/II clinical trial. Using this approach, a response rate of 30 % has been reported in non-small cell lung carcinoma (NSCLC), with no serious toxicities observed [151]. LY2157299 is a small molecule inhibitor which is selective for the kinase domain of the type 1 TGF-β receptor. LY2157299 is currently being evaluated in patients with metastatic malignancies.
7.3.2 IL-17
7.3.2.1 Overview
IL-17 is a proinflammatory cytokine produced by Th17 cells [152]. In addition to Th17 cells, IL-17 can also be produced by cells other than T helper cells, such as iNKT, CD8+ T, and γδ-T cells [153–155]. Since Th17 cells produce large quantities of IL-17A, most Th17-mediated effects are attributed to this cytokine. Many factors are required for the induction and stabilization of Th17 cells. Of these, TGF-β and IL-6 are the most crucial cytokines for its differentiation. IL-6 induces production of IL-21, which subsequently favors Th17 differentiation in an autocrine manner [156, 152]. These cells require CD40-induced IL-23 to maintain their Th17 phenotype in vivo. The differentiation of Th17 cells into IL-17-secreting cells requires the expression of the transcription factor ROR-γt [157]. It has been shown that Th17 cells are gradually increased in the tumor microenvironment during tumor development. In addition, Th17 cells have been found in patients with different tumors. The frequent association of raised IL-17 concentrations with negative prognosis links the increased systemic IL-17 concentrations with the later stages of cancer. Many factors released by the tumor cells and molecules secreted by tumor-infiltrating immune cells such as TGF-β, IL-6, prostaglandin E2 (PGE2), IL-21, IL-23, osteopontin, IL-1β, and TNF-α can play major roles in the induction of Th17 differentiation [158–161].
7.3.2.2 Th17 Differentiation in the Tumor Microenvironment
There are many sources for Th17 cells in the tumor microenvironment. Th17 cells found in the tumor microenvironment can either be migrated from the periphery or differentiated from naïve T cells under the influence of tumor microenvironmental factors. Th17 cells can traffic to tumors from the periphery under the influence of tumor microenvironmental chemokines such as RANTES and monocyte chemotactic protein-1 (MCP-1) [162]. In addition, high levels of chemokines CXCL12 and CCL20 have been found in tumor microenvironments, which could facilitate Th17 cell trafficking and migration into the tumor sites. Moreover, Th17 cells in the tumor microenvironment might clonally expand following stimulation by tumor-associated macrophages [163]. In addition, Th17 cells can be induced and differentiate in the tumor microenvironment [164]. It has become clear that IL-17 producing Th17 cells and Tregs share a common pathway. Although TGF-β favors differentiation of naïve T cells into Tregs, the simultaneous presence of both TGF-β and IL-6 promotes the differentiation of Th17 cells. Given the tight association of TGF-β and IL-6 with tumor incidence and progression, naïve T cells entering an established tumor are more likely to be exposed to conditions favoring Th17 differentiation. Upon stimulation with TGF-β and IL-6, CD8+ T cells not only lose their cytotoxic ability but are also induced to secrete IL-17 [165]. IFN-γ expressed by Th1 or CD8+ T cells inhibits angiogenesis and induces major histocompatibility complex I in tumor cells, thus favoring immune recognition and subsequent arrest of tumor growth [166]. In contrast, IL-17 favors angiogenesis and tumor growth; therefore, replacing IFN-γ with IL-17 in the tumor microenvironment may have severe consequences for immune recognition and surveillance.
7.3.2.3 Tumor-Promoting Functions of IL-17
Many functions of IL-17 in the tumor microenvironment contribute to tumor progression, besides their minor direct effect on the proliferation and survival of tumor cells [167]. The major pro-tumor role of IL-17 in cancer relies on its proangiogenic property of the surrounding endothelial cells and fibroblasts. For example, IL-17-overexpressing human cervical cancer cells and NSCLC cells show greater ability in developing tumors in immunocompromised mice compared with control cells with no IL-17 expression [168, 169]. In addition, IL-17 overexpression in fibrosarcoma cells enhances their tumorigenic growth in syngenic mice, primarily owing to the proangiogenic activity of IL-17. Moreover, the levels of Th17 cells were positively correlated with microvessel density in tumors [170]. By acting on stromal cells and fibroblasts, IL-17 induces a wide range of angiogenic mediators [171, 172] including VEGF. In addition, they markedly promote inflammatory and tumor angiogenesis [173]. IL-17 is able to upregulate VEGF production by fibroblasts and therefore promotes fibroblast-induced new vessel formation in the inflammatory microenvironment and tumors. The IL-17-VEGF loop that modulates angiogenesis includes another angiogenic factor, TGF-β. IL-17 induces VEGF, which in turn induces TGF-β followed by VEGF-mediated angiogenesis [174]. TGF-β enhances the VEGF receptivity of endothelial cells by increasing VEGF receptor expression [175]. IL-17 also induces IL-6 and PGE2 and enhances intercellular adhesion molecule (ICAM)-1 expression in fibroblasts. All these molecules were known to have a major role in angiogenesis and tumor invasion. IL-17 appears to stimulate production of IL-8 [176]. IL-8 signaling promotes angiogenic responses in endothelial cells, increases proliferation and survival of endothelial and cancer cells, and potentiates the migration of cancer cells and infiltrating neutrophils at the tumor site. Moreover, IL-17 was found to induce IL-1β and TNF-α in macrophages and cytokines which can further synergize with IL-17 to activate neutrophil-specific chemokine, thereby recruiting neutrophils to the site of inflammation [177].
One of the most important mechanisms underlying IL-17 regulation of inflammation which orchestrates the tumor microenvironment is through NF-κB signaling, the master regulator of the inflammation [178]. IL-17R signaling results in the activation of NF-κB and regulates the activities of extracellular-regulated kinase 1 (ERK1), ERK2, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinases [179, 180]. While the IL-17-mediated cytokine expression is regulated primarily by NF-κB, the same cytokines can further stimulate NF-κB-mediated transcription of themselves in tumor cells and tumor-associated stromal cells, thereby creating a sustained chronic inflammatory state within the tumor microenvironment. In support of this notion, enhanced cervical cancer growth elicited by IL-17 was associated with increased expression of IL-6 and macrophage recruitment to the tumor sites [169]. Therefore, IL-17 might also function through IL-6 to promote tumor development. Chemokines can stimulate or inhibit proliferation and chemotaxis of the blood vessel endothelial cells which serve the tumor. IL-17 has been shown to selectively enhance the production of angiogenic chemokines such as CXCL1, CXCL5, CXCL6, and CXCL8 from tumor cells and epithelial cells [168, 181]. In addition, IL-17 is also known to inhibit angiostatic chemokine secretion by fibroblasts [168]. Thus, IL-17 may shift the local biologic balance between angiogenic and angiostatic chemokines toward a predominance of angiogenic chemokines in order to enhance the net angiogenic activity.
7.3.2.4 Antitumor Functions of IL-17
Although IL-17 seemed to be a potential tumor-promoting cytokine, a considerable number of reports have described tumor-inhibitory effects of IL-17. Th17-polarized cells were found to be more effective than Th1 cells in eliminating large established tumors [182]. However, the Th17-mediated tumor responses were highly dependent on IFN-γ-based mechanisms. Indeed, the effects of Th17-polarized cells were completely abrogated by the administration of IFN-γ-depleting Ab and not by IL-17- or IL-23-depleting Abs. Adoptively transferred IL-17-secreting CD8+ T cells also enhanced antitumor immunity resulting in regression of B16 melanoma [183]. In addition, IL-17 has been shown to inhibit the growth of hematopoietic tumors such as mastocytoma and plasmocytoma by enhancing CTL activity [184]. Different mechanisms have been proposed for the IL-17 enhancement of tumor-specific CTLs. IL-17 has been shown to induce IL-6 from a variety of cells. Moreover, IL-17 stimulation can induce IL-12 production from macrophages [185]. Both IL-6 and IL-12 have been associated with induction of tumor-specific CTL. IL-17 promotes maturation of DC progenitors as indicated by increased expressions of co-stimulatory molecules, MHC-II antigens, and allostimulatory capacity [186]. This may lead to further improvement in T-cell priming by tumor cells producing IL-17. In addition, IL-17-transduced fibrosarcoma cells induced tumor-specific antitumor immunity by augmenting the expression of MHC class I and class II antigens [187]. These studies were focused on the effects of exogenous IL-17 in established mouse tumor cell lines. A recent demonstration shows that tumor growth in subcutaneous and lung tumor metastasis are enhanced in IL-17-deficient mice [188]. The effect is accompanied by reduced IFN-γ levels in tumor-infiltrating NK cells and T cells.
The evidence reviewed here demonstrates that IL-17-secreting Th17 cells can either stimulate or inhibit tumor growth and progression. The beneficial effects of IL-17 on upregulating host immune response may be present early in inflammation, but is eventually overcome by increasingly large tumor burden. Clearly, as discussed above, many of the inflammatory functions of IL-17 can benefit the tumor. The shift from beneficial inflammatory functions of IL-17 to a detrimental one may depend on the tumor type and inflammatory mediators in the tumor microenvironment. The pro-tumor vs. antitumor effects of IL-17 are thus functions of a number of combinations of all these IL-17-induced inflammatory mediators and, perhaps, mediators which counter-regulate IL-17 production as well, operating in tandem.
7.3.3 IL-23
7.3.3.1 Overview
IL-23 is a heterodimeric protein composed of two subunits IL-23p19 and IL-12p40. IL-23 is secreted by activated DCs and macrophages. IL-23 binds the IL-23R complex, composed of IL-23R and IL-12Rβ1. Upon binding IL-23, IL-12Rβ1, and IL-23R associate, marking the beginning of the IL-23 signal-transduction cascade [189]. IL-23 plays an important role in bridging innate and adaptive responses. Therefore, IL-23 has been described as a key cytokine promoting inflammation in peripheral tissues. The activity of IL-23 in the regulation of tumor immunity is just beginning to be elucidated [190].
7.3.3.2 Pro- vs. Antitumor Functions of IL-23
Belonging to the IL-12 family, IL-23 performs both pro-tumor and antitumor functions. IL-23 is spontaneously produced by TAM in several mouse tumor models. Tumor-secreted PGE2 enhances the production of IL-23 and IL-1β in macrophages and DCs while downregulating IL-12 production [191–193]. While IL-12 production is decreased, IL-23 production is increased in tumors [194]. PGE2, together with IL-23, favors the expansion of human Th17 cells from PBMCs; on the other hand, PGE2 enhances IL-17 production from memory CD4+ cells induced by IL-23 [161]. The involvement of IL-23 in the induction of Th17 was established when investigators showed that IL-23 promotes the production of IL-17 by activated T cells [195]. Although IL-23 is not involved in the initial differentiation of Th17 cells, it is crucial for the function, survival, and propagation of this T-cell population in the inflamed environment. In contrast to the antitumor role of IL-12, IL-23 upregulates inflammatory processes, including matrix metalloproteinase expression and angiogenesis and reduces infiltration and the function of CTLs, thus contributing to tumor growth [196]. Indeed, the mice lacking IL-23/p19 are completely resistant to carcinogen-induced tumor. The absence of tumor formation in these mice correlated with the absence of various markers including IL-17, GR-1+, and CD11b+ myeloid cells which are indicative of tumor-associated inflammation [196]. Recently, tumor-secreted lactic acid has been shown to activate the IL-23/Th17 pathway [159].
In contrast, IL-23-overexpressing tumors show reduced growth and metastasis [197–201]. The antitumor effects of IL-23 in these studies were found to be mediated through enhancement of CD8+ T-cell response. In addition, intra-tumoral injection of IL-23-overexpressing DCs resulted in a similar phenotype [201]. Artificial overexpression of IL-23 could induce potent antitumor immunity through various mechanisms. IL-23 can mediate myeloid infiltration consisting of DCs, macrophages, and granulocytes, which instead may contribute to the inhibition of tumor growth and boost an immune reaction to these immune-sensitive tumors. In addition, overexpression of IL-23 is likely to increase the systemic IL-23 levels that could lead to the growth and survival of memory CD8+ T cells.
7.3.4 IL-35
7.3.4.1 Overview
IL-35 is a recently discovered IL-12 family cytokine composed of an IL-12 p35 subunit and an IL-12 p40-related protein subunit, EBI3 [202]. IL-35 is not constitutively expressed in tissues and is produced mainly by Tregs. IL-35 induces the transformation of CD4+ effector T cells into Tregs, which in turn express IL-35 but lack the expression of conventional Treg markers such as Foxp3, TGF-β, and IL-10 (Treg35 cells) [203]. The Treg35 cells generated in vitro can prevent the development of autoimmunity in various mouse models [204–207]. Most recently, it has been shown that human Tregs express and require IL-35 for maximal suppressive function. Substantial upregulation of EBI3 and IL12A, but not IL10 and TGF-β, was observed in activated human Tregs compared with conventional T cells [208].
7.3.4.2 Pro-tumor Functions of IL-35
Evidence on the role of IL-35 in tumor immunity is beginning to emerge. IL-35 subunit EBI3 is expressed in Hodgkin lymphoma cells, acute myeloid leukemia cells, and lung cancer cells [209–211]. Small interfering RNA silencing of EBI3 in lung cancer cells inhibits cancer cell proliferation, whereas stable expression of EBI3 in lung cancer cells confers growth-promoting activity in vitro [211]. High EBI3 expression in human lung cancer cells was shown to be associated with poor prognosis [211]. Recently, IL-35-secreting Ag-specific Tregs have been observed in patients with prostate cancer [212]. Treg-derived IL-35 was shown to inhibit antitumor T-cell responses. In vitro generated Treg35 cells accelerate the development of B16 melanoma and prevent the generation of antitumor CD8+ T-cell responses [203]. In addition, T cells that secrete IL-35 and have suppressive functions can be induced in the tumor beds of melanoma and colorectal adenocarcinoma. Blockade of IL-35 has been shown to relieve suppression mediated by Tregs [212]. Forced expression of IL-35 in tumor cells leads to significantly increased tumorigenesis in mice. IL-35 in the tumor microenvironment significantly increased the numbers of CD11b+Gr1+ myeloid cells in tumors and subsequently promoted tumor angiogenesis [213]. Furthermore, IL-35 renders tumor target cells more resistant to CTL destruction.
7.3.5 IL-10
7.3.5.1 Overview
IL-10 is an important immunoregulatory cytokine produced by many cell populations. Due to its ability in inhibiting the production of IL-2 and IFN-γ by murine and human Th1 cells, IL-10 was initially named a cytokine synthesis inhibitory factor [214]. The function of IL-10 in cancer is enigmatic. Depending on the experimental model, IL-10 displays both immunosuppressive and immunostimulating activities. On the one hand, IL-10 promotes an antitumor CTL response leading to tumor regression. On the other hand, IL-10 induces immunosuppression and assists in the escape from tumor immune surveillance, hence promoting tumor growth.
7.3.5.2 IL-10-Mediated Immunosuppression in Cancer
The cellular sources of IL-10 are Th2, Treg, Tr1, and Th17 cells; however, cytotoxic CD8+ T cells can also produce IL-10, as can some subsets of DCs, macrophages, B cells, granulocytes, mast cells, keratinocytes, and epithelial cells. In addition, various cancer cells produce IL-10; among those are multiple myeloma, melanoma, human colon carcinoma, lung cancer, oral squamous cell carcinoma, renal cell carcinoma, non-Hodgkin lymphoma, and chronic lymphocytic leukemia [15, 215]. Circulating concentrations of IL-10 were raised in patients with different cancer types and were associated with adverse disease stage or with negative prognosis in those patients. It has been shown that serum levels of IL-10 increased in parallel to clinical disease progression in patients with metastatic melanoma, as well as colon cancer. Moreover, preoperative serum levels of IL-10 predicted the likelihood of colon cancer recurrence [215, 216]. IL-10 can be induced and sustained in the tumor microenvironment by a variety of cytokines. Macrophage-derived IL-6 has been shown to induce production of IL-10 by cancer cells. Similarly, IL-6 in association with TGF-β can induce IL-10 production in Th17 cells. However, TGF-β alone can induce IL-10, whereas IL-10 enhances the expression of TGF-β in a positive feedback circuit [217]. TNF-β promotes proinflammatory reactions while upregulating IL-10 in macrophages and monocytes as a negative feedback, thereby terminating the inflammatory response. In addition, IL-12 and IL-27 can also induce IL-10 production from T cells [99, 218].
Related posts:
 Prognostic Value of Innate and Adaptive Immunity in Cancers
Prognostic Value of Innate and Adaptive Immunity in Cancers
 Autophagy and Necroptosis in Cancer
Autophagy and Necroptosis in Cancer
 MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
 B Cells in Cancer Immunology: For or Against Cancer Growth?
B Cells in Cancer Immunology: For or Against Cancer Growth?
 Immunohistochemistry of Cancers
Immunohistochemistry of Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


