Fig. 14.1
Autophagy signaling pathway and antitumor therapeutic targets
14.2 Autophagy and Cancer
The role of autophagy in cancer is rather perplexing. It is widely postulated that the autophagic pathway is deregulated in tumor cells. Several proteins and pathways related to autophagy signaling are deregulated during cancer development [25, 71]. Cell lines derived from hepatic, pancreatic, and breast carcinoma exhibit low autophagic activity, as compared with normal cells from the same origin [25, 72]. Autophagic capacity is known to increase during the premalignant stages of pancreatic carcinogenesis and then decreases during the transition of pancreatic adenoma into adenocarcinoma, suggesting that a decreased autophagic activity possibly contributing to the malignancy of pancreatic cancer [73, 74]. A decrease in autophagic capacity is also observed during animal experimental carcinogenesis, where cells from preneoplastic liver nodules or primary hepatocellular carcinomas induced by chemical carcinogens showed a decreased autophagic capacity as compared to normal liver cells [74, 75]. In addition, Beclin-1 is found to be mono-allelically deleted in a high percentage of ovarian, breast, and prostate cancers (based on the 17q21 and gene mapping studies). It has been demonstrated to have a direct link between tumorigenesis and the disruption of autophagy [25]. PTEN deletions as well as the amplifications of both Class III PI3K and Akt are found in several cancers [76, 77].
The mTOR signaling pathway is constitutively activated in many tumor types. For example, the mTOR pathway is frequently found to be hyperactive in cancers such as breast cancer, suggesting that mTOR is an attractive target for cancer drug development and therapy [78–80]. The mTOR signaling network contains a number of tumor suppressor genes which includes PTEN, LKB1 (liver kinase B1), TSC1/2, and a number of proto-oncogenes such as PI3K, Akt, and eIF4E genes [81]. Cancer-related changes in pathways at the downstream of mTOR such as p70S6k and eIF4E are reported in breast carcinoma [82, 83]. In addition, malignant cell types undergo autophagic cell death when responding to anticancer agents and traditional herbs, indicating the potential utility of autophagic cell death induction in cancer therapy [13, 84, 85]. Autophagic cell death characterized by an increase in the number of autophagic vacuoles in the cytoplasm, followed by cell demise, has been observed in various diseases such as Alzheimer’s disease [86], Huntington’s disease [87–90], and Parkinson’s disease [91]. Therefore, manipulation of autophagy may provide an attractive strategy to increase the efficacy of cancer treatments, prevent cancer development, and limit tumor progression.
However, autophagy is divergent in nature in both tumor suppression and tumor progression [92]. Although the argument supports that if cells cannot activate autophagy, protein synthesis will predominate over protein degradation, and cellular growth continues (typical characteristic of tumor cells), there are some exceptional cases. For example, a study in human epidermoid lung carcinoma cells revealed that the autophagic pathway in response to nutrient deprivation is not downregulated when compared to their normal counterparts [93]. Human colon cancer cells which are able to survive for long period of time in the absence of nutrients have a high rate of autophagy activity [94]. Studies in colorectal cancer cells revealed that these cancerous cells harbor functional autophagic machinery to prolong cell survival during shortages of nutrients [95]. A recent study by Fuji and co-workers has also showed that strong LC3 expression in the peripheral area of pancreatic cancer tissue is correlated with poor outcome and short disease-free period [96]. Activated autophagy observed in pancreatic cancer cells is thought to be a response to factors in the cancer microenvironment, such as hypoxia and poor nutrient supply.
Autophagy has been identified as the key mechanism of cell survival in estrogen receptor-positive (ER+) breast cancer cells undergoing treatment with 4-hydroxytamoxifen (4-OHT) [97]. Antiestrogen therapy is the standard treatment for ER+ breast cancers which improves overall survival and provides chemoprevention [98, 99]. Unfortunately, approximately half of the women treated with antiestrogen therapy either do not respond or their breast cancer ultimately acquires resistance during treatment [100, 101]. Studies have shown that autophagy activity reduces the efficacy of chemotherapy and tamoxifen therapy in ER+ breast cancer cells [97, 102, 103], supporting the thesis that blocking autophagy signaling pathways may provide a new mechanism of anticancer therapy for resistant tumors.
In another example, electron microscopy examination of autophagic vesicles in melanoma tumors from 12 patients enrolled in a Phase II clinical trial of temozolomide and sorafenib therapy revealed that autophagic index (mean number of autophagic vacuoles per cell) is significantly higher in patients who derived little or no clinical benefit from the combination of temozolomide and sorafenib treatment. Patients who had stable disease or responded to therapy had low levels of autophagy in their tumors. These findings validate the emerging preclinical evidence that autophagy plays a critical role in resistance to chemotherapy. Results of this study indicate that pretreatment levels of autophagy can predict resistance to therapy. Patients with aggressive melanoma are more likely to have higher levels of autophagy in their tumor and therefore may respond to autophagy inhibition as a therapeutic strategy [104]. Hence, the divergent nature of autophagy has resulted in strategies for using pro-autophagics or autophagy inhibitors depending on the inherent nature of the cancer involved.
14.3 Autophagy Signaling Pathways and Therapeutic Strategies in Cancer
14.3.1 mTOR Signaling Pathway Inhibitors
Rapamycin, as the first prototype of an mTOR inhibitor, has a strong immunosuppressive property but poor aqueous solubility. Therefore, its utilization at doses capable of exerting anticancer effects is rather limited [105]. Nevertheless, trials utilizing rapamycin as a single-agent or combination therapy are still being carried out. In a recent Phase I study of rapamycin and sunitinib in patients with advanced non-small-cell lung cancer (NSCLC), combination of rapamycin and sunitinib is reported to be well tolerated and has warranted further investigation in Phase II trials [106].
Various rapamycin analogs have since been developed. Temsirolimus (CCI-779) is the first mTOR inhibitor approved by the US FDA for cancer treatment and is considered a first-line treatment for patients with advanced renal cell carcinoma (RCC) with poor prognostic features [107]. A great deal of clinical trials was carried out for this drug, mainly as combination therapy with other chemotherapy drugs. Generally, clinical activity is observed in patients with bone and soft tissue sarcoma given a combination of temsirolimus and cixutumumab [108]; in patients with metastatic adrenocortical carcinoma, the same combination therapy results in 40 % of patients achieving prolonged stable disease [109]. However, Phase I and II clinical trials with temsirolimus and sorafenib carried out in patients with metastatic melanoma did not produce sufficient activity to justify further use [110, 111]. In Phase II trial for metastatic colorectal cancer, temsirolimus has limited efficacy in chemotherapy-resistant KRAS-mutant disease [112].
Everolimus is another rapamycin analog which is already approved as an anticancer agent. Everolimus (RAD001; rapamycin derivative 001) is a hydroxyethyl ether derivative of rapamycin that has been developed for oral administration [113]. This drug was approved by FDA for use in a variety of cancers, including advanced RCC, advanced pancreatic neuroendocrine tumors, renal angiomyolipoma, and HER2-negative breast cancer. Everolimus, a derivative of rapamycin, is structurally similar to temsirolimus and binds to an intracellular protein, FKBP-12, forming a complex that inhibits the mTOR kinase. A recent Phase II trial showed that everolimus demonstrates efficacy and acceptable tolerability in patients with advanced endometrial cancer [114]. A randomized Phase II study indicates that combination therapy of everolimus with tamoxifen increases the clinical benefit rate (defined as the percentage of all patients with complete or partial response or stable disease at 6 months), time to progression (TTP), and overall survival compared with tamoxifen in postmenopausal women with aromatase inhibitor-resistant metastatic breast cancer [115]. In patients with advanced NSCLC, Phase I study showed that combination therapy with everolimus and erlotinib provides acceptable tolerability and disease control [116].
Ridaforolimus (deforolimus or AP23573) has been tested in Phase I and II clinical trials and has shown promising results in several tumor types including sarcoma [105, 117]. In a Phase II clinical study of ridaforolimus in 216 patients with advanced bone and soft tissue sarcomas, the overall clinical benefit response (CBR) was 28.8 % with a median progression-free survival of 15.3 weeks. Interestingly, the archival tumor protein markers analyzed were not correlated with CBR [118]. Ridaforolimus receives fast track and orphan drug status from the US FDA, as well as orphan status from the European Medicines Agency. The FDA is currently reviewing its registration for maintenance therapy in patients with sarcoma [119]. In another Phase II trial study on the efficacy and safety of single-agent ridaforolimus in patients with relapsed or refractory hematologic malignancies, results were unremarkable. Of the 52 patients evaluated, partial responses were noted in five subjects, while hematologic improvement/stable disease was observed in less than half of the patients [120].
PI3K-Akt-mTOR pathway is often constitutively activated in human tumor cells and thus has been considered as a promising drug target. NVP-BEZ235 is a potent imidazo(4,5-c)quinoline derivative that inhibits PI3K and mTOR kinase activities by binding to the ATP-binding cleft of these enzymes and induces G1 arrest [121]. Preclinical studies have suggested that NVP-BEZ235 is a potent dual PI3K/mTOR modulator with favorable pharmaceutical properties. For example, it inhibits VEGF-induced HUVEC cell proliferation and survival in vitro and VEGF-induced angiogenesis in vivo [122]. The compound also inhibits microvessel permeability in BN472 mammary carcinoma grown orthotopically in syngeneic rats, suggesting that this compound is potentially anti-angiogenic [122]. Deregulated angiogenesis and high tumor vasculature permeability are known VEGF-mediated characteristics of human tumors. In addition, NVP-BEZ235 is found to produce significant tumor growth inhibition in xenograft models of pancreatic cancers and breast cancer cells [123, 124]. Phase I/II clinical trials of NVP-BEZ235 in patients with advanced solid malignancies and breast cancer were completed, but reports on the safety and efficacy of this drug have yet to be published. Other ongoing trials either as a single-agent or combination therapy with other chemotherapy drugs in breast cancer, prostate cancer, leukemia, and other advanced solid tumors are listed in the NIH ClinicalTrials.gov website.
14.3.2 Pro-autophagics
Temozolomide is the first pro-autophagic cytotoxic drug used to overcome apoptosis resistance in cancer cells and was approved for use in glioblastoma multiforme (GBM) [125]. It demonstrates therapeutic benefits in patients with glioblastoma and has been evaluated for several types of apoptosis-resistant cancers [126]. Temozolomide is a prodrug, is a monofunctional alkylating agent, and is chemically related to dacarbazine. It is the 3-methyl derivative of the experimental anticancer drug, mitozolomide. The ability of temozolomide in inducing autophagic cell death is reported in various preclinical studies [127–130]. In addition, temozolomide also demonstrates pro-apoptotic activities in malignant melanoma cells [131]. In a systematic assessment of three randomized controlled trials addressing whether temozolomide holds any advantage over conventional therapy for high-grade gliomas, it was shown that temozolomide is an effective therapy for GBM. The drug prolongs survival, delays disease progression, and has a low incidence of early adverse events [132]. Similar outcomes were observed in a Phase II study involving erlotinib in combination with radiation therapy and temozolomide to treat GBM and gliosarcoma. Patients treated with the combination of erlotinib and temozolomide during and following radiotherapy have better survival than historical controls [133].
In a recent Phase II trial, patients with unresectable or multifocal glioblastoma, an upfront regimen of temozolomide and bevacizumab was well tolerated and provided a significant level of disease stabilization [134]. In patients with recurrent glioblastoma, either used as a single agent in a dose-intense schedule or in combination with other chemotherapeutic agents, temozolomide is proven to be well tolerated and safe [135–137]. In pediatric patients with recurrent solid tumors or brain tumors, low-dose temozolomide improves tolerability and is convenient as outpatient therapy [138]. Temozolomide in combination with vorinostat is also well tolerated in children with recurrent central nervous system (CNS) malignancies with myelosuppression [139]. However, good therapeutic effects are not observed in patients with NSCLC. In a current efficacy and safety study of temozolomide in a total of 31 pretreated patients with NSCLC, only two patients achieved partial response, and three had stable disease [140]. Moreover, the researchers pointed out that prolonged low daily doses of temozolomide produces minimal activity in patients with advanced NSCLC. Hence, more Phase II and III studies to characterize the efficacy of this drug in various cancers are definitely warranted.
Arsenic trioxide (ATO) has recently been introduced as part of a regimen in the therapy and management of acute promyelocytic leukemia (APL) [141]. It is now considered to be “the most biologically active single drug in APL” by a panel of international leukemia experts for the European LeukemiaNet. The combination of ATO and all-trans retinoic acid (ATRA) holds the promise to “replace conventional approaches for most, if not all, patients in the very near future” [142]. ATO is known to induce both autophagy and apoptosis depending on cell types; therefore, its role as an autophagy inducer remains largely uncertain. In some preclinical trials, ATO has induced the autophagic pathway in ovarian carcinoma cells and has synergized with everolimus to induce the cytotoxicity of ovarian cancer cells. The enhanced cytotoxicity is accompanied by the upregulation of Atg5-Atg12 conjugate and LC3-II, a hallmark of autophagy [143]. In another recent study, ATO induced the autophagic degradation of the BCR-ABL1 oncoprotein, known to cause chronic myeloid leukemia (CML) and Ph+ acute lymphoblastic leukemia (ALL) [144]. However, in other studies, in the presence or absence of ionizing radiation and in specific low concentrations, ATO induced apoptosis in MTLn3 cells, known to be highly malignant and resistant to both radio- and chemotherapy [145]. Interestingly, in human glioma cells, ATO induces both autophagy and apoptosis in vitro and in vivo, mediated by the inhibition of PI3K/Akt and activation of MAPK signaling pathway [146].
In a Phase I clinical study, ATO given concomitantly with radiation therapy in children with newly diagnosed anaplastic astrocytoma, glioblastoma, or diffuse intrinsic pontine glioma was safe and well tolerated by patients throughout the entire dose escalation [147]. ATO is also reported to be well tolerated when used in combination with temozolomide and radiotherapy in malignant gliomas [148] or when used in combination with bortezomib, high-dose melphalan, and ascorbic acid in multiple myeloma (MM) patients [149]. A Phase II study to evaluate the efficacy and feasibility of a sequential treatment consisting of induction and consolidation with ATO followed by autologous hematopoietic cell transplantation for relapsed APL revealed that ATO demonstrates outstanding efficacy. Of the 23 patients who underwent autologous hematopoietic cell transplantation with PML-RARα-negative PBSC graft, posttransplant relapse occurred only in three patients, and there was no transplant-related mortality. The 5-year event-free and overall survival rates were 65 % and 77 %, respectively [150]. Phase I/II/III clinical trials using ATO mostly as combination therapy with other chemotherapy drugs are currently ongoing for CML and APL.
14.3.3 Autophagy Inhibitors
The knowledge that autophagy plays a role as a cell survival pathway in response to therapeutic and cellular stresses in the tumor microenvironment (which is highly acidic and hypoxic) implies that autophagy may work in favor of cancer cells. Therefore, inhibition of protective autophagy may break the resistance mechanism for survival of the harsh tumor microenvironment and lead to cell death [151]. Since autophagy activities are known to differ according to stages of cancer, modulation of autophagy is postulated to enhance the efficacy of anticancer therapy. In a preclinical study, effects of imatinib, with or without different types of autophagy inhibitors, on human malignant glioma cells were carried out [152]. It is demonstrated that suppression of imatinib-induced autophagy by 3-methyladenine (3-MA) or siRNA against Atg5 (which inhibits autophagy at an early stage) attenuates the imatinib-induced cytotoxicity. On the other hand, inhibition of autophagy at a late stage by bafilomycin A1 or RTA 203 enhanced imatinib-induced cytotoxicity through the induction of apoptosis [152]. Thus, the authors have even suggested that therapeutic efficiency of imatinib for malignant glioma may be augmented by inhibition of autophagy at a late stage, which could help sensitize tumor cells to anticancer therapy [152].
The current autophagy inhibitors used in trials for human cancer are chloroquine (CQ) and hydroxychloroquine. Both drugs are widely used as antimalarials and have recently received attention as potential chemosensitizers in treating tumors when used in combination with cytotoxic chemotherapeutic agents [153–155]. CQ inhibits lysosomal acidification and therefore prevents autophagy by blocking autophagosome fusion and degradation [154, 156, 157]. A number of clinical trials are now revealing the promising role of CQ, an autophagy inhibitor, as a novel antitumor drug. For example, adding chloroquine to conventional treatment for GBM improves midterm survival of patients [158]. In a Phase I study involving patients with advanced NSCLC, hydroxychloroquine, with or without erlotinib, was safe and well tolerated, although the overall response rate was as low as 5 % [159]. Other trials on metastatic breast cancer, pancreatic cancer, RCC, NSCLC, and MM are currently ongoing. Table 14.1 summarizes the various drugs targeting the autophagy pathways and clinical trial stages based on published reports as well as ongoing trials listed in the NIH ClinicalTrials.gov website.
Table 14.1
Current therapeutic targets in the autophagy signaling pathways and clinical trial stages
Pathway | Therapeutic targets | Current drugs | Clinical trial stages (published reports)/type of cancer | Combined with | References |
|---|---|---|---|---|---|
mTOR signaling pathway | mTOR | Rapamycin (sirolimus) | Phase I: advanced NSCLC | Sunitinib | [106] |
Ongoing Phase I: NSCLC | BIBW 2992 | ||||
Ongoing Phase I: solid tumor | – | ||||
Ongoing Phase II: hepatocellular carcinoma | – | ||||
Temsirolimus (CCI-779) | Approved by FDA for advanced RCC (2007) | – | The US Food and Drug Administrationa | ||
Phase I: melanoma | Sorafenib | [111] | |||
Phase I: advanced adrenocortical carcinoma | Cixutumumab | [109] | |||
Phase II: melanoma | Sorafenib | [110] | |||
Phase II: bone and soft tissue sarcoma | Cixutumumab | [108] | |||
Phase II: colorectal cancer | Irinotecan | [112] | |||
Ongoing Phase I: advanced cancer | Vemurafenib | ||||
Ongoing Phase I: solid tumor | Erlotinib | ||||
Ongoing Phase I: advanced cancer | Bevacizumab and valproic acid | ||||
Ongoing Phase I: Hodgkin lymphoma | – | ||||
Ongoing Phase II: diffuse large B-cell lymphoma | Rituximab, cytarabine, cisplatin, and dexamethasone | ||||
Ongoing Phase II: head and neck cancer | – | ||||
Everolimus (RAD001) | Approved by FDA for advanced RCC (2009), advanced pancreatic neuroendocrine tumor (2011), renal angiomyolipoma (2012), and hormone receptor-positive, HER2-negative breast cancer (2012) | – | The US Food and Drug Administrationb | ||
Phase I: advanced NSCLC | Erlotinib | [116] | |||
Phase II: advanced breast cancer | Tamoxifen | [115] | |||
Phase II: advanced endometrial cancer | – | [114] | |||
Ongoing Phase II: advanced breast cancer | Exemestane | ||||
Ridaforolimus (deforolimus; AP23573) | Phase II: hematologic malignancies | – | [120] | ||
Phase II: advanced bone and soft tissue sarcoma | – | [118] | |||
PI3K/mTOR | NVP-BEZ235 (BEZ235) | Ongoing Phase I: advanced solid tumor, breast cancer, or renal cell carcinoma | Everolimus | ||
Ongoing Phase I: advanced solid tumor | – | ||||
Ongoing Phase I: acute leukemia | – | ||||
Ongoing Phase I: breast cancer | Capecitabine | ||||
Ongoing Phase I: advanced solid tumor | – | ||||
Ongoing Phase Ib: castration-resistant prostate cancer | Abiraterone acetate | ||||
Ongoing Phase I/II: castration-resistant prostate cancer | Abiraterone acetate and prednisone | ||||
Ongoing Phase I/II: advanced solid tumor | Everolimus | ||||
Ongoing Phase Ib/II: breast cancer | Paclitaxel | ||||
Ongoing Phase II: advanced pancreatic neuroendocrine tumor | Everolimus | ||||
Ongoing Phase II: perivascular epithelioid cell tumor | – | ||||
Ongoing Phase II: advanced pancreatic neuroendocrine tumor | – | ||||
Pro-autophagics | Temozolomide | Approved by FDA for GBM (2005) | Radiotherapy | [125] | |
Phase I: glioblastoma | Tipifarnib and radiotherapy | [137] | |||
Phase I: primary brain or spinal cord tumor | Vorinostat | [139] | |||
Phase I: solid tumor or brain tumor | Bevacizumab, vincristine, and irinotecan | [138] | |||
Phase I/Ib: glioblastoma | Lonafarnib | [136] | |||
Phase II: advanced NSCLC | – | [140] | |||
Phase II: GBM or gliosarcoma | Erlotinib and radiotherapy | [133] | |||
Phase II: glioblastoma | Bevacizumab | [134] | |||
Phase II: glioblastoma | – | [135] | |||
Ongoing Phase I: glioblastoma | BKM120 | ||||
Ongoing Phase I: acute leukemia | Veliparib | ||||
Ongoing Phase I/II: melanoma | Doxycycline and ipilimumab | ||||
Ongoing Phase I/II: melanoma | Decitabine and panobinostat | ||||
Ongoing Phase II: hepatocellular carcinoma | Veliparib | ||||
Ongoing Phase II: neuroblastoma | Bevacizumab and irinotecan | ||||
Ongoing Phase II: breast cancer | Veliparib | ||||
Ongoing Phase II: small cell lung cancer | Veliparib | ||||
Ongoing Phase III: glioblastoma | Lomustine | ||||
Ongoing Phase III: high-grade glioma | Interferon-α | ||||
Arsenic trioxide | Phase I: malignant glioma | Temozolomide and radiotherapy | [148] | ||
Phase I: infiltrating astrocytomas of childhood | Radiotherapy | [147] | |||
Phase II: multiple myeloma | Bortezomib, melphalan, and ascorbic acid | [149] | |||
Phase II: APL | – | [150] | |||
Ongoing Phase I: CML | Tyrosine kinase inhibitors | ||||
Ongoing Phase II: small cell lung cancer | – | ||||
Ongoing Phase II: APL | Tretinoin | ||||
Ongoing Phase II: APL | Gemtuzumab ozogamicin and ATRA | ||||
Ongoing Phase III: APL | ATRA | ||||
Ongoing Phase IV: APL | – | ||||
Autophagy inhibitors | Chloroquine | Ongoing Phase I: pancreatic cancer | Gemcitabine | ||
Ongoing Phase I/II: ductal carcinoma in situ | – | ||||
Ongoing Phase II: multiple myeloma | Bortezomib and cyclophosphamide | ||||
Ongoing Phase II: breast cancer | Taxane or taxane-like drugs | ||||
Hydroxychloroquine | Phase I: advanced NSCLC | Erlotinib | [159] | ||
Ongoing Phase I/II: RCC | Everolimus | ||||
Ongoing Phase I/II: NSCLC | Gefitinib | ||||
Ongoing Phase I/II: pancreatic cancer | Gemcitabine | ||||
Ongoing Phase I/II: colorectal cancer | FOLFOX and bevacizumab |
14.4 Mechanisms of Necroptosis
Necrosis is initially known as a passive and uncontrolled death process usually caused by physical or chemical insult. An irreversible drop in intracellular ATP and energy insufficiency lead to the morphological characteristics of organelle swelling, plasma membrane rupture, and spillage of cytoplasmic content [160, 161]. DNA in necrotic cells is usually degraded randomly, giving rise to a smear of DNA [162]. The cellular content leaks into the extracellular environment and is usually associated with inflammation [163, 164]. Release of cytokines and other factors from the necrotic cells and the secretion of pro-inflammatory cytokines from activated macrophages triggered by necrotic cells are thought to be responsible for the inflammatory response [165, 166]. Interestingly, in the past decade, studies have revealed necrosis as a form of regulated cell death, executed through a mechanism termed necroptosis or programmed necrosis [167, 168]. Necroptosis can be stimulated via a class of death receptors including TNFR1, TNFR2, TRAIL-R, and Fas. Upon binding to their agonists, these death receptors can induce cells toward either survival or death. Depending on the circumstances, the induction of cell death may be either apoptosis or necroptosis. However, the exact mechanisms that dictate the cellular decision to undergo apoptosis or necroptosis remained largely unknown.
TNF-α can be massively generated during hyperinflammatory shock, accumulated upon infection or produced primarily by macrophages. It induces apoptosis in many cells, while triggering necrosis in some [161, 169]. In necroptosis, TNF-α binds to the extracellular portion of the death receptors and triggers downstream signaling pathway by forming complex I with proteins containing a death domain, such as TNF-receptor-associated death domain (TRADD), receptor-interacting protein kinase 1 (RIP1), and several E3 ubiquitin ligases, such as TNF-receptor-associated factor 2/5 (TRAF2/5), cIAP-1, and cIAP-2. Ubiquitination of these proteins is important for the regulation of the activity of complex I and impacts the outcome of the cell survival [170]. The ubiquitination and phosphorylation states of RIP1 determine whether it functions as a pro-survival molecule or a kinase promoting cell death. RIP1 is a member of the RIP family exhibiting a homologous N-terminal kinase domain and has recently emerged as an essential mediator of cellular stress and cell death. RIP1 is polyubiquitinated by TRAF2/5, cIAP-1, and cIAP-2 at the 63rd position of lysine (K63) [171, 172]. K63-linked ubiquitination of RIP1 by cIAP-1/cIAP-2 is known to inhibit TNF-α-induced apoptosis [173, 174].
Complex I is crucial for activating the NF-κB and mitogen-activated protein kinase (MAPK) pathways. cIAPs direct the formation of polyubiquitin chains on RIP1, allowing it to interact with TGF (transforming growth factor)-β-activated kinase 1/TAK-1-binding protein 2/3 (TAK1/TAB2/3) complex. TAK1 activates the IκB kinases (IKK) complex and, in turn, phosphorylates IκBα. When IκBα is degraded by the proteasome, it allows NF-κB to translocate to the nucleus and activate its target genes in a pro-survival manner. Inhibitors of NF-κB are known to facilitate TNF-α-induced necrotic cell death, suggesting that NF-κB suppresses the necrotic cell death pathway [175]. On the other hand, deubiquitination of RIP1 could inhibit NF-κB pathway, leading to cell death pathways. Cylindromatosis (CYLD) blocks the activation of NF-κB by cleaving K63-linked polyubiquitin chains, and its deubiquitinating activity on RIP1 facilitates the direct interaction of RIP1 with caspase-8 and initiation of cell death [176, 177]. Knockdown of CYLD inhibits TNF-α-induced necroptosis, which indicates that the deubiquitination of RIP1 is an important step in TNF-α-induced necroptosis [178]. CYLD also interacts directly with TRAF2, an adaptor molecule involved in signaling of the TNF/nerve growth factor family receptors. TRAF2, an E3 ligase, has been demonstrated to be essential for TNF-α-induced necroptosis, as TRAF2−/−cells are resistant to TNF-α-induced necroptosis [175].
When RIP1 is deubiquitilitized by CYLD, RIP1 can dissociate from complex I and is released into the cytoplasm, forming complex II with FADD, RIP3, and caspase-8. If the conditions are apoptotic competent, TNF-α stimulation induces the sequential protein complexes, complex I and complex IIa, leading to the activation of NF-κB and apoptosis, respectively [168, 179]. However, proteolytical cleavage of RIP1 by caspase-8 during TNF-induced apoptosis abolishes NF-κB activation and enhances pro-apoptotic signaling through the TRADD-FADD interaction [180]. Cleavage of RIP3 by caspase-8 induces caspase-dependent apoptosis [181]. However, if the apoptotic machinery is deficient or when the apoptosis pathway is blocked by pan-caspase inhibitors such as Z-VAD-FMK, or caspase-specific inhibitors such as cytokine response modifier A (CrmA) or in cells deficient in FADD or caspase-8, triggering TNFR1 results in necrosis [182–186]. This process involves the formation of complex IIb, consisting of mainly RIP1 and RIP3. It appears that FLICE-inhibitory protein (FLIP), together with caspase-8, is recruited to FADD and the formation of this complex is dominant for inhibiting apoptosis [187]. Cellular FLICE-inhibitory protein (c-FLIP) is known as a crucial inhibitor of death receptor-mediated apoptosis by interfering with caspase-8 activation at the death-inducing signaling complex (DISC) signaling [188]. Due to its structural similarity to caspase-8 and caspase-10, c-FLIP can bind to FADD and inhibit complete caspase-8 processing and activation [189]. However, the involvement of FADD, caspase-8, and FLIP in complex IIb remains unclear.
RIP3 has an N-terminal kinase domain and a C-terminus lacking a death domain or CARD motif. RIP3 binds RIP1 through this unique C-terminal segment to inhibit RIP1- and TNF-receptor-1-mediated NF-κB activation [190]. However, the interaction between RIP1 and RIP3 via the RIP homotypic interaction motif (RHIM) domain is required for necroptosis [191]. RHIMs of RIP1 and RIP3 mediate the assembly of heterodimeric functional amyloid signaling complex which is ultrastable [192]. Mutations in the RHIMs of RIP1 and RIP3 which render them defective in interactions compromise kinase activation and necroptosis in vivo, indicating the crucial role of RHIM in necroptosis [192]. Mutations of RHIMs in RIP1 or RIP3 block the formation of necrosomes and protect cells from necroptosis [190]. RIP3 acts upstream to phosphorylate RIP1, which in turn mediates downstream RIP3 phosphorylation. Phosphorylation of RIP3 is essential in necroptosis, but the exact mechanism remains unclear [191]. Both RIP3 and the kinase activity of RIP1 are essential for stable formation of the RIP1-RIP3 pro-necrotic complex, which critically controls downstream ROS production [191]. RIP3 is essential in necroptosis induced by various stimuli, and RIP3 knockdown leads to a notable inhibition of necroptosis [191, 193]. Cells with low levels of RIP3 expression are resistant to necroptosis, but transfection of these cells with the RIP3 gene enables them to undergo necroptosis when the apoptotic pathway is blocked, clearly highlighting RIP3 as an essential mediator in TNF-α-induced necroptosis [194]. In addition to TNF-α, IFN-γ also induces an NF-κB-dependent transcriptional response that is cytoprotective. However, in mammalian cells deficient in NF-κB signaling, IFN-γ promotes mitochondrial ROS accumulation, loss of mitochondrial membrane potential, and necroptosis [195]. The necroptosis signaling pathway is illustrated in Fig. 14.2.
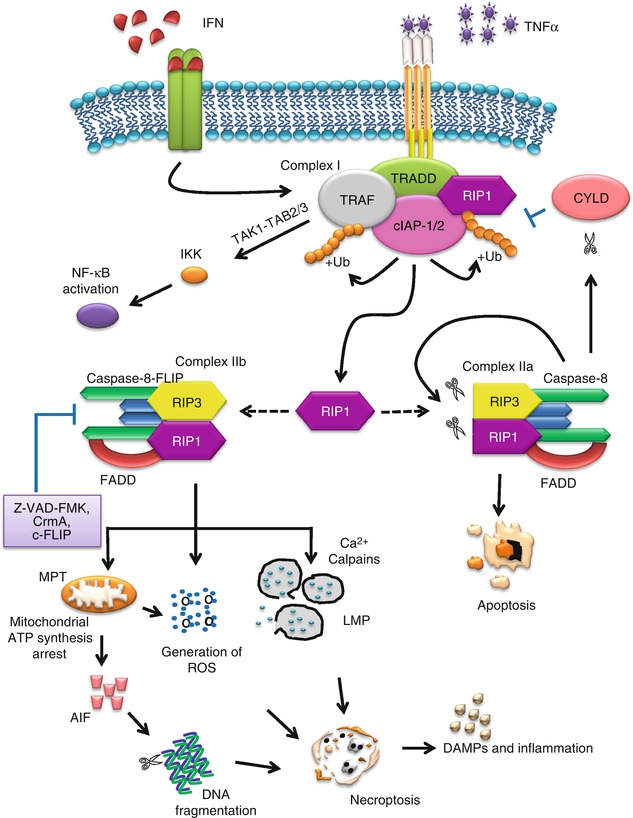
Fig. 14.2
Necroptosis signaling pathways
Necroptosis shows identical subcellular events with necrosis and secondary necrosis. The cellular disintegration phases are characterized by lysosomal membrane permeabilization, mitochondrial hyperpolarization, oxidative burst, and eventually plasma membrane permeabilization; however, the kinetics and timing may be different [196]. A number of events have been implicated and proposed to contribute to the downstream events in necroptosis. One important downstream event is the production of ROS which acts as an executioner of necroptosis [168]. The ROS is implicated to play an important role in necroptosis; in addition, RIP, TRAF2, and FADD are crucial in mediating ROS accumulation in TNF-induced necroptotic cell death [175]. This was based on the observation that in TNF-induced necroptosis, the cellular ROS level was significantly elevated in wild type, but not in RIP(−/−), TRAF2(−/−), and FADD(−/−) cells [175].
Interestingly, RIP3 has been reported to interact with several metabolic enzymes including glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), and glutamate dehydrogenase 1 (GLUD1) [197]. PYGL plays a key role in using reserved glycogen as an energy source and catalyzes the rate-limiting step in the degradation of glycogen by releasing glucose-1-phosphate. On the other hand, GLUL is a cytosolic enzyme catalyzing the condensation of glutamate (Glu) and ammonia to form glutamine (Gln). Gln transfers into the mitochondria to function as an energy substrate. GLUD1 is a mitochondrial matrix enzyme that converts Glu to α-ketoglutarate. GLUL and GLUD1 are essential for the use of amino acid Glu or Gln as substrates for adenosine triphosphate (ATP) production by means of oxidative phosphorylation [197]. Taken together, these enzymes increase substrates for oxidative phosphorylation, which is a major source of ROS in the cell. RIP3-deficient cells have reduced ROS production downstream of TNF-α signaling [191]. Zhang and co-workers postulated that RIP3 activation of all these enzymes results in an increased energy metabolism and subsequent ROS production [197].
Nicotinamide adenine dinucleotide phosphate oxidases (NADPH) are a family of enzymes specifically important in ROS production and have been implicated in TNF-α-induced necroptosis. For example, TNF treatment induces the formation of a signaling complex containing TRADD, RIP1, Nox1, and the small GTPase Rac1. RIP1 is shown to be essential for Nox1 recruitment, and activation of Nox1 is implicated in ROS production [198]. Other NADPH oxidases, such as Nox1, Nox2, Nox3, and Nox4, are also shown to be upregulated in the presence of TNF-α [198–200]. In addition, riboflavin kinase (RFK), a TNFR1-binding protein, functionally couples TNFR1 to NADPH oxidase. RFK binds to both the TNFR1 death domain and p22phox, the common subunit of NADPH oxidase isoforms and triggers TNF-induced ROS production [201]. Both RFK and the NADPH oxidases are found to be crucial for downstream ROS production [198, 201].
ROS are thought to act by oxidizing MAP kinase phosphatases (MKPs) whose normal function is to downregulate the c-Jun N-terminal kinase (JNK) signaling pathway [202]. Sustained JNK activation is required for Cyt c release and caspase-3 cleavage in apoptosis as well as necroptosis [202]. In necroptosis, JNK is activated in the downstream of RIP1 and TRAF2 [203, 204]. ROS and JNK appear to form a loop to enhance necroptosis as JNK also affects the mitochondrial function and produces ROS [205]. JNK activation is required for mitochondrial depolarization, AIF translocation, and subsequent cell death in PARP-1-hyperactivated cells [204].
Activation of phospholipase A2 (PLA2) and lipoxygenase pathways are also known to contribute to the TNF-α-induced necrotic death [206]. Cytosolic PLA2 (cPLA2), a subfamily of PLA2, is an intracellular enzyme that hydrolyzes arachidonate-containing phospholipids and facilitates the release of arachidonic acid. Arachidonic acid is the main substrate for lipoxygenase, which further catalyzes the conversion of the fatty acids into hydroperoxides [163]. Overexpression of cPLA2 sensitizes TNF-α-resistant cells to TNF-α-induced necrosis, emphasizing the role of cPLA2 in necroptosis [207, 208]. Lipid peroxidation leading to disruption of organelle and plasma membranes are key features of necrosis.
The mitochondrion has been implicated downstream of RIP1. Mitochondrial synthesis of ATP requires ADP transport from cytosol into mitochondria by the inner mitochondrial membrane ADP/ATP carrier adenine nucleotide translocase (ANT) [209]. ADP/ATP exchange depends on transition between two conformational states of ANT. In the cytosolic state (c-state), the hydrophilic loop of the ANT nucleotide-binding site faces the cytosol, while in the matrix state (m-state), this binding site faces the matrix [209, 210]. The interaction of ANT with cyclophilin D (CYPD) and voltage-dependent anion channel (VDAC) is important in regulating the mitochondrial permeability transition pore (MPTP); in addition, CYPD is an important regulator of MPTP [211]. Z-VAD-FMK is found to block the ability of ANT to transport cytoplasmic ADP, thereby inducing a massive ATP depletion in the mitochondria [212]. The inhibition of ADP/ATP exchange coincides with the loss of interaction between ANT and CYPD as well as with the inability of ANT to adopt the cytosolic conformational state, which prevents Cyt c release and subsequently necroptosis [212].
The release of cytosolic Ca2+ and overactivation of calpains are also thought to play important roles in necroptosis. Yamashima and co-workers postulated that excessive Ca2+ overload leads to calpain-mediated lysosomal disruption with releases of cathepsins B and L [213]. Cathepsin B is shown to be involved in caspase-independent cell death induced by death receptor ligands [213]. Lysosomal membrane permeability (LMP) is associated with activation of PLA2, causing the production of ROS [214]. LMP is also known to activate mitochondrial permeability transition (MPT), leading to cell death. The opening of MPTP accounts for the MPT resulting in disruption of the inner mitochondrial transmembrane potential (Δψm) during cell death [215–217]. Oxidative stress can serve as a facultative inducer of MPTP opening; moreover, ROS are potent inducers of MPTP opening [218–220]. The induction of MPT, which increases mitochondrial membrane permeability, causes the mitochondria to become further depolarized, resulting in the abolishment of Δψm as well as allowing the release of ROS and other molecules such as AIF and necrotic danger-associated molecular patterns (DAMPs) into the cytosol.
AIF is a FAD-dependent oxidoreductase that has a vital role in oxidative phosphorylation [221]. After a caspase-independent cell death insult, AIF is cleaved by calpains and/or cathepsins to yield truncated AIF (tAIF), the pro-apoptotic AIF form (~57 kDa) [222, 223]. The tAIF relocates from the mitochondria to the cytosol and nucleus, where it associates with histone H2AX in the nucleus, through its C-terminal proline-rich-binding domain (PBD, residues 543–559). This interaction generates an active DNA-degrading complex with cyclophilin A, leading to chromatin condensation and DNA fragmentation, as observed in necroptotic cells [224].
Interestingly, necroptosis induced by high doses of the alkylating DNA-damaging agent N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) is found to be regulated by the kinase RIP1 and executed by the activation of PARP-1, Ca2+-dependent calpain Cys proteases, and the pro-apoptotic Bcl-2 member Bax [225]. MNNG treatment induces a PARP-1 hyperactivity that leads to calpain activation. Calpains generate tBid, which redistributes from the cytosol to mitochondria, where it regulates Bax activation. Once activated, Bax provokes mitochondrial damage and tAIF mitochondrial release. The tAIF relocalizes to the nucleus, associates with H2AX and cyclophilin A, and subsequently induces chromatinolysis [226]. PARP-1 is a nuclear enzyme activated by DNA strand breaks and plays a key role in repairing DNA damage. PARP-2, the closest homolog to PARP-1, has been identified as one of the essential regulators of necroptosis by a genome-wide siRNA screen study [178]. PARP activation is also found to play a critical role in glutamate-induced necroptosis [227]. The mechanisms of PARP-induced mitochondria dysfunction in necroptosis remain to be explored.
Necroptotic cells spill their contents which contain DAMPs. DAMPs can trigger inflammation by activating pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors, and retinoic acid-inducible gene I (RIG-1)-like receptors [228]. DAMPs are intracellular molecules that have inflammation-inducing capacities when released from cells, resulting in the activation of macrophages and subsequently the inflammation processes [229].
14.5 Necroptosis and Possible Therapeutic Targets in Cancer
Necroptosis is found to occur during the early phases of T-cell clonal expansion, indicating that this mode of cell death may be involved in the regulation of the immune system [230]. In addition, virus-infected cells, which are resistant to apoptosis, are found to be highly sensitive to necroptosis, indicating that it may serve as an alternative mechanism of cell death other than apoptosis [231]. In acute lymphoblastic leukemia (ALL), necroptosis cell death can be induced to overcome the glucocorticoid resistance of ALL cells. Resistance to the initial phase of chemotherapy, in particular poor response to glucocorticoids, is a strong predictor of adverse outcome for childhood ALL. In a clinical study using primary leukemia cells from patients with very high-risk disease, obatoclax mesylate, a Bcl-2 antagonist, and rapamycin increased RIP1 activity and restored the response to dexamethasone by inducing a type of cell death morphologically consistent with necroptosis [232]. In addition, necroptosis appears to cause cancer cell death as a response to several anticancer treatment strategies, clearly indicating the role of this cell death in cancer [233–236].
Defects in necroptosis or variations of necroptosis-related genes may contribute to the pathological process of human malignancies, based on several observations and studies.
Deubiquitination of RIP1 by CYLD is important for the formation of complex II, leading to either apoptosis or necroptosis. Tumors carrying the mutated CYLD(C/S) [catalytically inactive form of CYLD that mimics the identified mutations of CYLD in human tumors] exhibit a faster growth, are poorly differentiated, have robust angiogenesis activity, and are aggressive tumors [237]. Both cIAP-1 and cIAP-2 may promote cancer cell survival by functioning as E3 ubiquitin ligases that maintain constitutive ubiquitination of the RIP1 adaptor protein [174]. cIAP-1 and cIAP-2 directly ubiquitinate RIP1 and induce constitutive RIP1 ubiquitination in cancer cells which then associates with the pro-survival kinase TAK1 and suppresses apoptosis [174]. Immunohistochemical analysis of cIAP-1 and cIAP-2 in archival bladder specimens revealed that both cIAP-1 and cIAP-2 expression are significantly increased in bladder cancer compared with normal bladder urothelium. Nuclear cIAP-1 expression is strongly correlated to bladder cancer stage, tumor grade, and tumor recurrence suggesting the possibility of using cIAP-1 as a marker in bladder cancer prognosis [238]. Furthermore, X-IAP, cIAP-1, and cIAP-2 are found to be highly expressed in chronic lymphocytic leukemia samples [239–242]. Thus, the IAPs should be an attractive antitumor strategy. In addition, RIP1 polyubiquitination by TRAF2/TRAF5 at the position of K63 inhibits TNF-α-induced apoptosis [171, 172] and TRAF2 knockdown by siRNA radiosensitizes cancer cells via a reduced NF-κB activation, suggesting that TRAF2 may also be an attractive target for anticancer activity [243].
Necroptosis in cancer cells is induced by various approaches including administration of alkylating DNA-damaging agents [244] and the application of photodynamic therapy through which photosensitizing compounds accumulated in tumor cells generate ROS following excitation with light from various spectra [245, 246]. Necroptosis induction is speculated to be useful in cancers which are resistant to the apoptotic effects of chemotherapy. One preclinical study on shikonin, a naturally occurring naphthoquinone, has demonstrated that induction of necroptosis by the compound is able to overcome resistance to cancer drugs mediated by P-glycoprotein, Bcl-2, and Bcl-XL in cancer cell lines [236]. All these results indicate that necroptosis could be a potent therapeutic strategy for the treatment of cancer. The further exploration of the necroptosis signaling pathway will be important to identify strategies and novel antitumor drugs which can be brought forward to the human clinical trials.
14.6 Crosstalk in Apoptosis, Autophagy, and Necroptosis
Functional relationships between apoptosis and autophagy are gaining much interest, as both cell deaths are not mutually exclusive. Perturbations in the apoptotic machinery, such as caspase inhibition, have been reported to induce both autophagic cell death and necroptosis [247, 248]. Inhibition of autophagy in cancer cells results in an accelerated cell death that manifests the hallmarks of apoptosis including chromatin condensation, MOMP, and activation of caspases [249]. In some cases, mixed phenotypes of both autophagy and apoptosis are detected in response to common stimuli [156, 249]. Studies in a variety of experimental systems indicate that autophagy cell death is likely to be cell type dependent. Autophagy can delay the onset of apoptosis, following starvation, DNA damage, and hemodynamic stress [13]. For example, 1-day fasting causes liver autophagy in rats, but when starvation is prolonged for a few days, hepatocytes succumb to apoptosis [250]. Similarly, hematopoietic cell lines withdrawn from growth factor first activate autophagy and eventually apoptosis [7]. Studies have also demonstrated that certain compounds have the ability to trigger both apoptosis and autophagy cell deaths simultaneously in cancer cells [251, 252]. Blocking of one pathway will trigger the activation of another [253]. Researchers have also hypothesized that there are factors (either external or internal) that may affect the preferential shunting into either biochemical cascades that will ultimately result in either apoptosis or autophagic cell death [254].
Crosstalks between autophagy and apoptosis exist at multiple levels because both pathways share mediators and pathway regulators. Several signals and pathways involved in autophagy are in common with apoptosis. Starvation and oxidative stress can trigger both apoptosis and autophagy. Bcl-2 proteins function to inhibit both apoptosis and autophagy, providing another clue to the interplay between both processes. Beclin-1, the essential autophagy protein and haplo-insufficient tumor suppressor, interacts with several cofactors such as Ambra1, Bif-1, and UVRAG to activate the lipid kinase Class III PI3K and induce autophagy [255]. In normal conditions, Beclin-1 is bound to and inhibited by Bcl-2 or the Bcl-2 homolog Bcl-XL, well-characterized apoptosis regulators, which involve an interaction between the BH3 domain in Beclin-1 and the BH3-binding groove of Bcl-2/Bcl-XL. BH3-only proteins can competitively disrupt the interaction between Beclin-1 and Bcl-2/Bcl-XL to induce autophagy. Nutrient starvation can stimulate the dissociation of Beclin-1 from its inhibitors, either by activating BH3-only proteins (such as Bad) or by posttranslational modifications of Bcl-2 (such as phosphorylation) that may reduce its affinity for Beclin-1 and BH3-only proteins [255]. Anti-apoptotic Bcl-2 family members participate in the inhibition of autophagy, whereas the pro-apoptotic BH3-only proteins participate in the induction of autophagy.
A recent finding suggests a link between autophagy and the extrinsic apoptotic pathway mediated by p62/SQSTM1. Autophagy is recently known to be responsible in selective degradation of polyubiquitinated proteins via sequestosome-1 (SQSTM1), which encodes for p62 protein. p62 interacts with LC3 via its LC3 interacting region (LIR). Recent studies indicate that p62 is recruited to damaged mitochondria via binding to ubiquitinated outer mitochondrial membrane proteins, suggesting that p62 may serve as an autophagy receptor for ubiquitinated proteins and damaged mitochondria [256–258]. In addition to its role in autophagy, p62 mediates a cell’s decision to undergo apoptosis or survival through its organization of signaling complexes in the cytoplasm [257, 259, 260]. Upon cytokine stimulation, p62 activates the NF-κB pathway, which subsequently induces the pro-survival genes, such as anti-apoptotic and cell proliferation genes and induces the expression of inflammatory genes such as cytokines, chemokines, and adhesion molecules [260–263]. However, p62 is also found to activate caspase-8 in the extrinsic apoptosis pathway, resulting in the initiation of apoptosis and cell death [259].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


