Fig. 1
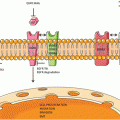
A simplified overview of platinum resistance in lung cancer. Pt platinum, CTR1 copper transporter 1, GSH glutathione, BCRP breast cancer resistance protein
Table 1
Tumor factors contributing to resistance to commonly used chemotherapeutic agents in lung cancer
Agents | Factors |
|---|---|
Platinum | Drug accumulation Copper transporters (CTR1, ATP7B) Na+, K+ ATPase Breast cancer resistance protein Drug detoxification Glutathione DNA repair Nucleotide excision repair (ERCC1) Metallothioneins |
Taxanes | Class III β-tubulin Expression levels Mutations Hypoxia inducible factor Histone deacetylase 6 Mitotic spindle checkpoint |
Etoposide | Drug uptake Topoisomerase II alpha Rad51 Mitogen-activated protein kinases (MAPKs) ERK1, ERK2 Metallothionein Non-homologous end-joining repair |
Gemcitabine | Human equilibrative nucleoside transporter 1 (hENT-1) Ribonucleotide reductase M1 and M2 (RRM1 and RRM2) |
Vinorelbine | Polo-like kinases (PLKs) Class III β-tubulin Delta2 α-tubulin RLIP76 Stathmin (oncoprotein 18) Mitotic spindle checkpoint |
Pemetrexed | Drug transport Proton-coupled folate receptor Folate receptor-α |
Once platinum drugs enter the cell, they must form DNA adducts, which induce a cascade of signaling transduction pathways that lead to activation of p53-dependent and p53-independent cell death [34]. In NSCLC patients who were treated with concurrent cisplatin/XRT, low cisplatin-DNA-adduct staining in buccal cells was associated with worse survival [35]. While DNA adduct level is a critical determinant of cisplatin cytotoxicity, increased DNA adduct repair decreases the apoptotic process [34]. Nucleotide excision repair is the major pathway for platinum adduct repair. The excision repair cross-complementation group 1 (ERCC1) protein is involved in nucleotide excision repair pathway and its expression correlated with cisplatin resistance in NSCLC [36]. There was a major interest in further developing ERCC1 as a potential biomarker for platinum sensitivity in NSCLC when Olaussen and colleagues demonstrated that ERCC1-negative NSCLC specimens determined by IHC correlated with significantly longer OS following adjuvant chemotherapy [37]. There have been several studies with conflicting results since then. For example, a phase III trial designed to determine the influence of ERCC1 mRNA on tumor response following chemotherapy failed to show a correlation between ERCC1 expression and treatment response [38]. Furthermore, Friboulet and colleagues failed to validate the predictive effect of ERCC1 protein expression by IHC in two independent phase III trials (the National Cancer Institute of Canada Clinical Trials Group JBR.10 and the Cancer and Leukemia Group B 9633 trial from the Lung Adjuvant Cisplatin Evaluation Biology project) [39]. The authors concluded that currently available ERCC1 antibodies do not have adequate discrimination for clinical decision making regarding cisplatin-containing treatment regimens in patients with NSCLC, which requires the specific detection of the unique functional isoform of ERCC1 [39]. Furthermore, the results from the randomized international phase III trial of ERCC1 and RRM1 expression-based chemotherapy versus gemcitabine/carboplatin demonstrated no significant difference between the experimental arm and the control arm in PFS or OS [40]. Therefore, utility of ERCC1 as a predictive marker for platinum-based chemotherapy appears to be limited at this moment.
Metallothioneins are low molecular weight proteins that are involved in zinc homeostasis and may also be involved in chemotherapy binding and detoxification. Increased metallothionein expression was noted in some cisplatin-resistant NSCLC [5] and SCLC [41] cell lines. In patients with SCLC receiving cisplatin-based therapy, metallothionein expression determined by IHC correlated significantly with short survival [42].
Taxanes
The taxanes such as paclitaxel and docetaxel stabilize microtubules inhibiting the disassembly process, resulting in cell death due to large numbers of spurious asters forming throughout the cytoplasm [43]. Taxane-resistant NSCLC cell lines had significantly increased expression of class III β-tubulin [44–46]. Furthermore, the expression level of endogenous hypoxia inducible factor (HIF)-1 alpha appears to modulate taxane sensitivity in vitro by influencing the conformation and dynamics of microtubules [47]. In NSCLC cell lines that are resistant to taxanes, there was increased microtubule instability [48]. In addition, histone deacetylase 6 (HDAC6) decreased microtubule stability and antagonized the effect of paclitaxel on NSCLC cells. On the other hand, the farnesyl transferase inhibitor lonafarnib blocked the effect of HDAC6 on tubulin and was synergistic with paclitaxel [49]. The specimens from NSCLC patients demonstrated that a majority of the tumor samples expressed class II and class III tubulins, although the percentage of positive cells varied significantly among tumors [50]. NSCLC patients whose tumors expressed low levels of class III beta-tubulin isotype had a better response rate, longer PFS, and OS. However, this variable was not predictive in patients receiving regimens without tubulin-binding agents [51]. A multivariate analysis demonstrated that low-level class III beta-tubulin expression was independently associated with PFS and OS, suggesting that the expression levels of class III beta-tubulin in tumor cells are predictive of response to therapy and patient outcome in patients with NSCLC receiving paclitaxel-based chemotherapy [51].
In addition to the expression levels of beta-tubulin, the mutations in beta-tubulin gene may also predict response to the taxanes. Beta-tubulin mutations in exons 1 or 4 were found in 33 % of NSCLC patients, and none of these patients had an objective response to paclitaxel treatment [52]. In the same study, median survival was 3 months for the patients with beta-tubulin mutations and 10 months for the patients without mutations [52]. Overall, the available data suggest that elevated expression and mutations in class III β-tubulin may predict resistance to taxanes in lung cancer, offering an opportunity for personalization.
Another potential mechanism of taxane resistance in lung cancer is through dysregulation of the mitotic spindle checkpoint. The mitotic spindle checkpoint, which blocks segregation of abnormal chromosomes, is often defective in human lung cancer cell lines [53]. Anti-microtubule agents such as taxanes activate the mitotic spindle checkpoint [53]. In NSCLC cell lines, impairment of the mitotic spindle checkpoint was associated with marked reduction in the ability of docetaxel to induce apoptosis, compared to cell lines with an intact mitotic spindle checkpoint [53].
Etoposide
Etoposide is an inhibitor of topoisomerase II enzyme that prevents re-ligation of the DNA strands leading to potentially lethal DNA breakage [54]. There is relatively little information available on the potential role of decreased drug uptake in etoposide resistance, although etoposide uptake is significantly higher in sensitive SCLC cell lines than in more resistant NSCLC lines [55]. The nonionic detergent Tween-80 increased etoposide uptake and cytotoxicity in NSCLC cells [56]. On the other hand, some etoposide-resistant cell lines have significantly increased level of cholesterol [57], which is thought to increase cell membrane rigidity.
As expected, SCLC cell lines are generally more sensitive to topoisomerase II inhibitors than NSCLC cell lines are [55, 58]. Kasahara and colleagues reported that nuclear topoisomerase II activity was twofold higher in SCLC cells than in NSCLC cells [55]. On a similar note, the expression of topoisomerase II-α mRNA and the protein levels in lung cancer cell lines were lower in the resistant variants compared to the sensitive variants [59]. Other etoposide-resistant SCLC cells derived from a patient who developed acquired resistance after initial response to etoposide demonstrated reduced topoisomerase II unknotting activity and reduced topoisomerase II-α expression [60]. In clinical tumor specimens, there was a significantly greater topoisomerase II-α expression by IHC in SCLC than in NSCLC [61, 62], and topoisomerase II-α IHC expression decreased significantly in SCLC tumors after therapy with etoposide [62]. Overall, these data suggest that reduced expression of topoisomerase II-α may increase resistance to etoposide.
Rad51 protein expression may play a role in etoposide resistance . In SCLC cells, resistance to etoposide correlated with protein levels of RAD51. Also, aberrant RAD51 gene expression altered both the efficacy of etoposide and repair of etoposide-induced DNA breaks [63]. Rad51 was expressed in 41 % of clinical NSCLC tumor samples but it did not correlate with resistance to etoposide [36]. Hence, the role of Rad51 in etoposide resistance will need further investigation in clinical setting.
ERK1 and ERK2 are examples of mitogen-activated protein kinases (MAPKs), and are downstream from Ras, Raf and MEK in the Ras pathway. Some etoposide-resistant SCLC cell lines had markedly increased MAPK activity [64]. Etoposide is less effective in hypoxic tumor cells [65]. A hypoxic environment activated the ERK pathway and increased resistance of NSCLC cells to etoposide. Conversely, inhibiting the ERK pathway reversed the hypoxia-induced resistance [65].
Other potential mechanisms for etoposide resistance in lung cancer include increased metallothionein expression and non-homologous end-joining (NEJH) repair. In addition to increased metallothionein expression seen in some cisplatin-resistant cell lines as discussed in Sect. 1, metallothionein expression also correlated with resistance to etoposide [66]. Exposure of lung cancer cell lines to cadmium or zinc increased metallothionein synthesis and increased resistance to etoposide [66]. NHEJ is a pathway that repairs double-strand breaks in DNA without the need for a homologous template. DNA-dependent protein kinase (DNA-PK) plays a crucial role in this pathway [67]. The efficacy of etoposide and etoposide-induced double strand breaks in SCLC cell lines varied with expression of DNA-PK proteins, suggesting a role for NHEJ repair in etoposide resistance [63].
Gemcitabine
Gemcitabine is a deoxycytidine analogue that, following uptake through nucleoside transporters, undergoes complex intracellular conversion to gemcitabine diphosphate and triphosphate that are important for cytotoxic effects [68]. Human equilibrative nucleoside transporter 1 (hENT-1) plays a role in cellular uptake, and hENT-1-deficient cells were demonstrated to be resistant to gemcitabine [69]. Liposome encapsulation may enhance gemcitabine uptake and cytotoxicity [70]. One NSCLC study reported that pretreatment hENT-1 expression did not directly correlate with tumor response or survival following gemcitabine-based chemotherapy, but only 16 % of the tumor specimens expressed hENT-1 by IHC [69]. In a separate study, none of the NSCLC patients who lacked hENT-1 expression determined by IHC responded to gemcitabine-based therapy [71].
Ribonucleotide reductase M1 (RRM1) encodes the regulatory subunit for ribonucleotide reductase which is a target of gemcitabine [72]. Ribonucleotide reductase plays an important role in cell growth and DNA repair [72]. Over-expression of RRM1 mRNA [72, 73] and genetic variations in RRM1 gene [74] correlated with resistance to gemcitabine in NSCLC cells. Bexarotene which counteracts RRM1 gene amplification [75] and RRM1 siRNA [76] both decreased resistance to gemcitabine. Furthermore, a significantly higher RRM1 mRNA expression was found in SCLC cell lines compared to NSCLC cells [77]. In clinical setting, NSCLC patients who were treated with gemcitabine alone or in combination with a platinum drug, RRM1 mRNA expression levels inversely correlated with tumor response [72, 78], time to progression [79] or survival [78–81]. Furthermore, RRM2, a related factor, also correlated inversely with response. Patients whose tumors had high expression of both RRM1 and RRM2 had significantly lower response rates, shorter time to progression and OS, compared to those whose tumors had low expression of both [82]. RRM1 expression appears to be a promising biomarker for gemcitabine-based therapy in NSCLC. However, a recent phase III study comparing ERCC1 and RRM1 expression-based chemotherapy with gemcitabine/carboplatin in advanced NSCLC demonstrated that there was no statistically significant difference in PFS and OS between the experimental arm and the control arm [40]. Further investigation is warranted.
Vinorelbine
Vinorelbine is an amphiphilic Vinca alkaloid with superior activity in NSCLC compared with other drugs in the same category [83]. It exerts its antitumor activity by binding to tubulin and inhibiting microtubule assembly, thereby preventing cell mitosis [84].
Stuckler and colleagues reported a potential role of RLIP76, a non-ATP binding transport protein, in facilitating the efflux of vinorelbine in NSCLC [83]. They demonstrated that RLIP76 catalyzes the transport of vinorelbine in a saturable manner that is dependent on vinorelbine and ATP [83]. Furthermore, threefold overexpression of RLIP76 in NSCLC and SCLC confers increased resistance to vinorelbine by decreasing intracellular vinorelbine concentration [83].
Polo-like kinases (PLKs) play a role in mitotic entry, spindle pole function and cytokinesis [85]. Expression of PLK 1 was elevated in NSCLC, and inhibiting it disrupted microtubule polymerization while potentiating the efficacy of vinorelbine [86]. NSCLC patients who had low class III β-tubulin mRNA expression in their tumors had significantly longer time to progression and OS than patients with high expression of class III β-tubulin following treatment with vinorelbine/cisplatin [79, 87]. Low Delta2 α-tubulin expression was also associated with significantly longer OS in advanced NSCLC patients treated with cisplatin/vinorelbine [87]. Conversely, when NSCLC patients received cisplatin/vinorelbine in adjuvant setting after undergoing surgical resection of their tumors, a greater benefit of the therapy was seen in patients with high versus low class III β-tubulin. Furthermore, the adjuvant therapy appeared to overcome the negative prognostic effect of high β-tubulin [88]. It is unclear why there is a discrepancy between adjuvant and metastatic setting in the influence of tubulin expression on vinorelbine efficacy.
Stathmin (oncoprotein 18) is a protein that plays an important regulatory role in tubulin dynamics. Transfection of the gene into lung cancer cells increased sensitivity to vinca alkaloids [89]. However, in patients with advanced NSCLC treated with vinorelbine plus cisplatin, time to progression was shorter in patients with high stathmin than with low stathmin mRNA expression [79]. Hence, its role in resistance remains unclear.
Similar to docetaxel as discussed in section “Taxanes”, impairment of the mitotic spindle checkpoint was also associated with significant reduction in the ability of vinorelbine to induce apoptosis, compared to cell lines with an intact mitotic spindle checkpoint [53].
Pemetrexed
Pemetrexed is one of the newer agents that is commonly used to treat advanced non-squamous NSCLC based on a phase III study by Scagliotti and colleagues that showed survival differences based on histologic type [90]. In this study, OS was superior for pemetrexed/cisplatin versus gemcitabine/cisplatin in patients with non-squamous NSCLC only [90]. Like all other chemotherapeutic agents, virtually all tumors develop resistance to pemetrexed.
The proton-coupled folate receptor [91], the reduced folate carrier [91, 92] and the folate receptor-α [93] all appear to play a role in transport of pemetrexed. As discussed above, pemetrexed is more active in non-squamous carcinomas than in squamous cell carcinomas [90], possibly related to the fact that adenocarcinomas in particular have significantly higher expression of folate receptor-α compared to squamous cell carcinomas [94]. Cytotoxicity due to pemetrexed occurs by inhibiting thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyltransferase (GARFT). Increased expression of these enzymes was associated with reduced cytotoxicity of pemetrexed in NSCLC cell lines [95]. Moreover, a recent meta-analysis evaluated the predictive value of TS and reported that NSCLC patients with lower TS expression could potentially benefit from pemetrexed-based chemotherapy [96]. High extracellular folate pools also markedly reduced pemetrexed cytotoxicity [97]. Further clinical investigation is warranted.
Multidrug Resistance Protein (ABCC, ABCB)
The factors that confer resistance to one agent may render tumors resistant to several other agents [98]. Alternating multiple agents with different mechanisms of action including cisplatin and paclitaxel does not improve clinical outcome in NSCLC [99]. The multidrug resistance (MDR) phenotype can be caused by transporters of the multidrug resistance protein (MRP) family (also known as ABCC) or MDR P-glycoprotein family (also known as ABCB).
A high proportion of SCLC [100, 101] and NSCLC [100, 102] cell lines express MRP mRNA, with greater MRP protein and/or mRNA expression in NSCLC than in SCLC cell lines [103, 104]. MRP expression is associated with decreased cellular drug accumulation of cisplatin [105], paclitaxel [106] and other agents [107]. In both SCLC and NSCLC cell lines, MRP mRNA or protein expression correlated significantly with resistance to vinca alkaloids [102, 103, 105, 107, 108], etoposide [100, 103, 104, 108], docetaxel [109], paclitaxel [106], gemcitabine [76] and cisplatin [103, 105]. However, an association between MRP expression and resistance was not seen in some cell lines [110–112]. In clinical specimens, MRP mRNA and/or protein by IHC was found in 32–100 % of NSCLC tumor specimens [100, 113–115]. MRP expression was significantly higher in more differentiated tumors than in less differentiated tumors [113] and in squamous cell carcinomas than in other NSCLCs [115]. MRP expression was also reported in SCLC clinical tumor samples [101].
The clinical value of tumor MRP expression remains uncertain. Response rates to platinum-based combinations were significantly lower in patients with SCLC whose tumors express MRP1 [116, 117] or MRP2 [118] compared to those with tumors that do not express these factors. Furthermore, MRP1 expression in SCLC tumors was significantly higher at relapse after treatment with cisplatin/etoposide compared to untreated tumors [116], suggesting that chemotherapy may upregulate expression of MRP. Likewise, in autopsy NSCLC tumor specimens, mRNA expression levels of MRP3 [119] and MRP5 [120] were significantly higher in patients who had been exposed to platinum drugs ante mortem than in patients who had not received platinum agents.
In advanced NSCLC, response rates to cisplatin/irinotecan were higher and survival was longer in patients with some MRP2 host genotypes than with other genotypes [121]. However, there was lack of correlation between MRP IHC expression and response to platinum-based combinations in other studies [21, 122]. In a different study involving NSCLC patients, MRP mRNA expression only correlated negatively with tumor response only in adenocarcinomas, and not in squamous cell carcinomas [123]. Furthermore, tumor MRP1 or MRP2 IHC expression did not correlate with survival in patients with resected NSCLC receiving adjuvant cisplatin plus a vinca alkaloid or etoposide [124]. Overall, preclinical data support a role for MRP in resistance to several types of chemotherapy. However, clinical data remain inconclusive. Further study is necessary.
Like MRP, MDR/P-glycoprotein may also render tumors resistant to chemotherapy by transporting drugs out of cells. In NSCLC cells, increased MDR1 mRNA and/or protein expression levels were associated with resistance to vinca alkaloids [102, 125–128], etoposide [125, 127], and taxanes [125–127, 129, 130]. MDR1/P-glycoprotein expression did not correlate significantly with sensitivity to platinum drugs [5, 110, 125, 126] or intracellular platinum accumulation [5, 110]. Some NSCLC and SCLC cell lines transfected with the MDR1 gene had augmented sensitivity to gemcitabine, and this augmented sensitivity was reversed by the P-glycoprotein inhibitor verapamil [112]. MDR1 gene overexpression was also seen in SCLC cell lines selected for resistance by exposure to paclitaxel [106] or etoposide [131, 132]. P-glycoprotein expression correlated with HIF-1 alpha expression in NSCLC cell lines [133] and in resected NSCLC tumors [134]. Its expression was higher in lung adenocarcinoma cells under hypoxia [133], but was reduced in tumors of patients who had nitroglycerin patches applied to improve tumor blood flow and oxygenation prior to surgical resection [134].
In clinical setting, MDR1 mRNA and/or P-glycoprotein were expressed in 11–32 % of chemo-naïve specimens [62, 135–138], but were expressed in 61 % of tumors that had been treated with chemotherapy [137]. However, MDR1 expression in NSCLC did not correlate with histology or with clinical characteristics [135]. In SCLC, MDR1 expression was seen in 13–60 % of tumor biopsy samples [62, 101, 138].
With MDR1/P-glycoprotein, there is stronger evidence of an association of expression with clinical outcome in SCLC than in NSCLC. In SCLC, there was a negative correlation between expression of P-glycoprotein and clinical outcome in patients treated with cisplatin-etoposide [116, 117, 138–141]. In addition, P-glycoprotein expression was significantly increased in tumors previously exposed to therapy compared to the expression in untreated tumors [62, 116]. Efficacy of cisplatin/etoposide in SCLC patients also correlated with MDR1 host polymorphisms, with a significantly better chemotherapy response in patients with the 3435 CC genotype (exon 26) compared with those who had both 3435 CT and TT genotypes [142].
In NSCLC , there was a significant correlation of tumor P-glycoprotein IHC expression with response in two studies using platinum and paclitaxel [143, 144]. Similar to the findings in SCLC, the MDR1 3435 CC host genotype was associated also in NSCLC with a better response to cisplatin-vinorelbine compared with the combined 3435 CT and TT genotypes [145]. However, in a number of other studies involving patients with advanced NSCLC [21, 138, 146], P-glycoprotein expression by IHC did not correlate with response to cisplatin-based regimens [21, 138, 146] that included vinca alkaloids [21, 138, 147], taxanes [21] or gemcitabine [21]. Furthermore, host MDR1 C3435T polymorphisms did not correlate with outcome in NSCLC patients who were treated with cisplatin/docetaxel [148].
P-glycoprotein antagonists have been assessed in both NSCLC and in SCLC. The P-glycoprotein antagonist verapamil enhanced paclitaxel accumulation and vinorelbine efficacy in P-glycoprotein overexpressing lung cancer cells [111]. The epidermal growth factor receptor (EGFR) inhibitor gefitinib also reversed P-glycoprotein mediated taxane resistance in NSCLC cell lines [130, 149], but did not translate to improvement in efficacy of chemotherapy in randomized clinical trials involving NSCLC patients [150, 151]. A phase II study investigating cyclosporine A, another P-glycoprotein antagonist, in combination with paclitaxel in NSCLC patients suggested a possible positive impact of the cyclosporine on paclitaxel efficacy [152]. However, when cyclosporine was added to etoposide plus cisplatin in treatment-naive NSCLC, there was no evidence of clinical benefit [153]. Similarly, while hydroxyurea is thought to reverse MDR1-associated resistance in vitro [154], combining paclitaxel with hydroxyurea in previously-treated patients with advanced NSCLC did not appear to improve clinical outcome [155].
Based on the above, there is strong preclinical evidence of an association between MDR1/P-glycoprotein expression and resistance to several agents in lung cancer cells, but the clinical evidence is inconclusive for an association with outcome especially in NSCLC.
Conclusion
Despite rapid advances in diagnostic technology and better understanding of lung tumorigenesis which have led to development of targeted agents, chemotherapy continues to remain as the backbone of treatment for both SCLC and NSCLC. There are several chemotherapeutic agents with different mechanisms of action being currently used to treat lung cancer. However, virtually all tumors develop resistance to all these agents. Persistent investigation to understand the molecular mechanisms of chemotherapy resistance and identification of predictive biomarkers for chemotherapy sensitivity are necessary. Improved understanding of resistance mechanism at the molecular level may offer opportunities to combine chemotherapeutic agents with molecularly targeted agents, which may be a promising strategy to overcome chemotherapy resistance and to optimize therapy for lung cancer patients.
References
1.
Feldman DR, Bosl GJ, Sheinfeld J, Motzer RJ (2008) Medical treatment of advanced testicular cancer. JAMA 299(6):672–684. doi:10.1001/jama.299.6.672 PubMed
2.
Gately DP, Howell SB (1993) Cellular accumulation of the anticancer agent cisplatin: a review. Br J Cancer 67(6):1171–1176PubMedCentralPubMed
3.
Andrews PA, Howell SB (1990) Cellular pharmacology of cisplatin: perspectives on mechanisms of acquired resistance. Cancer Cells 2(2):35–43PubMed
4.
Shellard SA, Fichtinger-Schepman AM, Lazo JS, Hill BT (1993) Evidence of differential cisplatin-DNA adduct formation, removal and tolerance of DNA damage in three human lung carcinoma cell lines. Anticancer Drugs 4(4):491–500PubMed
5.
Kawai H, Kiura K, Tabata M, Yoshino T, Takata I, Hiraki A, Chikamori K, Ueoka H, Tanimoto M, Harada M (2002) Characterization of non-small-cell lung cancer cell lines established before and after chemotherapy. Lung Cancer 35(3):305–314, S0169500201004305 [pii]PubMed
6.
Bungo M, Fujiwara Y, Kasahara K, Nakagawa K, Ohe Y, Sasaki Y, Irino S, Saijo N (1990) Decreased accumulation as a mechanism of resistance to cis-diamminedichloroplatinum(II) in human non-small cell lung cancer cell lines: relation to DNA damage and repair. Cancer Res 50(9):2549–2553PubMed
7.
Kim ES, Lee JJ, He G, Chow CW, Fujimoto J, Kalhor N, Swisher SG, Wistuba II, Stewart DJ, Siddik ZH (2012) Tissue platinum concentration and tumor response in non-small-cell lung cancer. J Clin Oncol 30(27):3345–3352. doi:10.1200/JCO.2011.40.8120 PubMedCentralPubMed
8.
Ishida S, Lee J, Thiele DJ, Herskowitz I (2002) Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A 99(22):14298–14302. doi:10.1073/pnas.162491399162491399 [pii]PubMedCentralPubMed
9.
Song IS, Savaraj N, Siddik ZH, Liu P, Wei Y, Wu CJ, Kuo MT (2004) Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol Cancer Ther 3(12):1543–1549, 3/12/1543 [pii]PubMed
10.
Chen HH, Yan JJ, Chen WC, Kuo MT, Lai YH, Lai WW, Liu HS, Su WC (2012) Predictive and prognostic value of human copper transporter 1 (hCtr1) in patients with stage III non-small-cell lung cancer receiving first-line platinum-based doublet chemotherapy. Lung Cancer 75(2):228–234. doi:10.1016/j.lungcan.2011.06.011 PubMedCentralPubMed
11.
Ishida S, McCormick F, Smith-McCune K, Hanahan D (2010) Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 17(6):574–583. doi:10.1016/j.ccr.2010.04.011, S1535-6108(10)00150-9 [pii]PubMedCentralPubMed
12.
Stewart DJ, Issa JP, Kurzrock R, Nunez MI, Jelinek J, Hong D, Oki Y, Guo Z, Gupta S, Wistuba II (2009) Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res 15(11):3881–3888. doi:10.1158/1078-0432.CCR-08-2196 PubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


