Fig. 6.1
Potential roles of FoxP3+ T cells and Th17 cells in tumors. (a) FoxP3+ T cells are made in the thymus as naïve-type FoxP3+ T cells, which migrate to lymphoid tissues. These FoxP3+ T cells can become the memory type after activation in secondary lymphoid tissues. Induced FoxP3+ T cells with memory-type FoxP3+ T cells and Th17 cells are made from naïve CD4+ T cells. FoxP3+ T cells suppress effector T cells and other immune cells and decrease tissue inflammation. Th17 cells produce IL-17 to induce inflammatory responses. FoxP3+ T cells and Th17 cells can trans-differentiate into other T-cell subsets such as Th1 and T-FH cells in appropriate cytokine and antigen priming conditions. (b) FoxP3+ T cells can promote tumor growth by suppressing antitumor immune responses at early and late stages. On the other hand, Th17 cells can induce immune responses that lead to eradication of tumor cells in a manner similar to other effector, CD8+, and γδ T cells. (c) In inflammatory conditions, FoxP3+ T cells and Th17 cells have the potential to play different roles. Th17 cells cause inflammation in tissues; hence, inflammatory tumors are formed and stimulated to grow. FoxP3+ T cells suppress the function of Th17 cells and other inflammatory T cells, leading to suppression of the tumorigenic process in inflamed tissues
6.2 Diversity of Tumor Microenvironments and Tumor Tissue Factors
Tumor microenvironment is highly heterogeneous, depending on tumor types and sites of formation. Together with tumor cells, fibroblasts, myofibroblasts, endothelial cells, mast cells, and other tissue cells make up tumors. Moreover, immune cells are an important component of tumors and are mainly composed of T cells, B cells, innate lymphoid cells, and myeloid cells. Tumor-associated myeloid cells are heterogeneous as well and contain immature and mature myeloid cells. Myeloid-derived suppressor cells (MDSC) are immature myeloid cells and highly enriched in tumors [10]. MDSC are composed of heterogeneous myeloid cells at various different stages. Compared to mature myelocytes such as dendritic cells (DCs) and macrophages, MDSC do not highly express cytokines, co-stimulatory molecules, and MHC class molecules. Therefore, they poorly support antitumor effector T-cell responses. Moreover, MDSC express various molecules that dampen immune responses. MDSC produce peroxynitrite for nitration and nitrosylation of many proteins in the tumor environment [11, 12]. A major target protein for nitration and nitrosylation is TCR, which becomes ineffective at activating T cells after the modifications [13]. They also express Arg1, inducible nitric oxide synthase (iNOS), and TGF-β1, among others [14]. Tumors also harbor many macrophages, which can be made from MDSC or myeloid progenitor cells [15]. Dendritic cells express indoleamine 2,3-dioxygenase (IDO) to regulate available tryptophan [16]. Other immune cells such as mast cells, NK cells, CD8+ T cells, and B cells are frequently found in many tumor types.
The tumor environment is low in both oxygen and pH. Tumor cells rapidly divide and therefore vigorously consume oxygen supplied via blood vessels. Tumor cells mainly utilize the aerobic glycolysis pathway to generate energy [17]. This can accumulate lactic acid and protons, leading to low extracellular pH [18]. The most common pH range in tumors is 6–6.5. The low acidic tumor environment leads to immune cell anergy. For example, cytotoxicity and cytokine secretion by CD8+ T cells are impaired at the low pH range [19].
Cells in the tumor microenvironment produce various cytokines and growth factors [20]. Some of these factors are drained into lymphatic vessels and form tumor-associated microenvironmental milieu in lymph nodes. If tumors have tumor-specific or tumor-associated antigens, these antigens are drained or transported into lymph nodes and presented to T cells via antigen-presenting cells (APCs). Effector and regulatory T cells can be made during this antigen priming process. The cytokine milieu is critical in determining the fate of differentiating T cells in tumor-draining lymph nodes. Again, the type and amount of cytokines and other factors produced in tumors are highly diverse among tumor types. Expression of IL-1α, IL-1β, IL-6, IL-11, and TNF-α was observed in colon carcinoma, colon adenoma, ovarian cancer, and gastric cancer [21–27]. IL-2 and IL-15 are expressed in melanoma. IL-10 and TGF-β are expressed in myeloma, colon cancer, lung cancer, and mammary carcinoma [28, 29]. Expression of IL-17, IFN-γ, and IL-4 has been observed in certain tumor types [30–32]. Expression of M-CSF, GM-CSF, and IL-3 has been observed as well [33–35]. These tumor-derived hematopoietic cytokines regulate myeloid cell-mediated inflammation and affect T-cell activity in tumors. Chemokines such as CXCL chemokines (CXCL1, 3, 6, 8, 10, and 12) and CCL chemokines (CCL1, 2, 5, 17, 25, and 28) are expressed in various tumor types [36–39]. Growth and angiogenic factors such as VEGF, EGF, and HGF are broadly expressed in a number of cancer types [40, 41]. The cell types producing these factors are not limited to tumor cells but can be from various cell types in tumors. For example, tumor-associated macrophages produce both inflammatory and immunosuppressive cytokines such as IL-1, IL-6, IL-10, and TGF-β [42].
T-cell receptor (TCR) activation signals are modified by the signals from co-stimulatory and co-inhibitory molecules, which are expressed by tumor cells and tumor-associated APC [43]. These molecules include B7-1, B7-2, programmed cell death-1 ligand (PD-L1), PD-L2, ICOS-L, B7-H2, B7-H3, B7-H4, and B7-H6. Among these, PD-L1-PD and B7-1/2-CTLA-4 play important roles in the formation of Tregs in tumor microenvironments [44–46]. Moreover, TNF receptor family members such as OX40, GITR, 4-1BB, and CD40 are expressed in tumors and regulate antitumor immune responses [47, 48].
Inflammatory mediators are produced in tumors. Cyclooxygenase-2 (COX-2) is highly expressed in malignant tumors [49, 50]. COX-2 expression is induced in hypoxic conditions or by cytokines and growth factors [51]. COX-2 generates prostaglandin H2 from arachidonic acid, which is processed to generate major inflammatory mediators such as PGD2, PGE2, PGI2, and TXA2. These mediators regulate angiogenesis and various aspects of inflammatory responses in tumors [49].
Some tumor types are under the influence of microbe-associated molecular pattern (MAMP) receptor ligands if tumors are formed in barrier tissues such as the intestine or in patients infected with pathogens. In mucosal tissues, decreased barrier functions due to tumorigenesis or preexisting inflammation can lead to bacterial invasion and induction of inflammatory responses. Furthermore, tumors that are associated with infection by papillomavirus (uterine cervical carcinoma), hepatitis B virus (hepatocellular carcinoma), Epstein-Barr virus (Burkitt’s lymphoma), human T-cell leukemia virus (adult T-cell leukemia), or herpes virus (Kaposi’s sarcoma) would be influenced by viral MAMPs. MAMPs and DAMPs activate Toll-like receptors (TLRs) [52]. TLR activation can induce tissue inflammation that promotes cancer [53]. MYD88 signaling is also required for activation of dendritic cells for proper formation of effector T cells. Without proper MYD88 signaling, Th2 cells ineffective in antitumor immunity can be made [54]. TLR signaling can work together with STAT3 and Notch pathways to activate MAPK and NFkB, which promote the survival and proliferation of tumor cells [55].
Retinoic acid is an anticancer agent. Retinoic acids such as all-trans retinoic acid (ATRA) and 9-cis RA are produced from retinol (vitamin A) by retinol metabolizing enzymes such as ADH and RALDH [56]. Epithelial cells and APCs in the intestine highly express these enzymes [57]. RALDH2 expression is induced during immune responses to increase the concentration of RA available in local tissue environments. Inflamed tissues or tumors are low in expression of RA-producing RALDH but are high in expression of RA-catabolizing CYP26 [58, 59]. In sum, the tumor microenvironment is made of highly diverse factors. Some are from tumor cells, while others are from tissue cells and immune cells. These factors have profound effects on T cells in tumors and associated lymphoid tissues as discussed in detail later in this chapter.
6.3 Generation of Tregs and Th17 Cells
FoxP3+ Tregs are made in the thymus as natural FoxP3+ T cells. They are also induced in the periphery from naïve CD4+ T cells. In addition, IL-10-producing Tregs (Tr1 cells) are made from naïve CD4+ T cells. Tregs produce suppressive cytokines such as IL-10, IL-35, and TGF-β [60–62]. These Tregs play critical roles in preventing autoimmune diseases. Tregs are generally made whenever effector T cells are formed during immune responses. This is important to limit the potentially inflammatory activities of effector T cells.
Induction of effector T cells and Tregs occurs mainly in secondary lymphoid tissues. One reason for this is that naive CD4+ T cells that become effector T cells and Tregs migrate mainly to secondary lymphoid tissues. However, memory/effector T cells can trans-differentiate into each other at any tissue sites upon antigen priming (Fig. 6.1a). Th1 cells are the most readily made effector T cells from naïve CD4+ T cells. IL-12, a cytokine produced from DCs, promotes the generation of Th1 cells. Th2 cells are made when IL-4 is abundant. Th17 cells are generated when IL-6, TGF-β, and other inflammatory cytokines are present during T-cell priming. MAMPs and TLR activation in tissues promote the generation of Th17 cells. Th1 cells are efficient in the promotion of cell-mediated immunity through production of IFN-γ Th17 cells that are effective at inducing inflammatory conditions through producing IL-17. A number of inflammatory cytokines, neutrophil-attracting chemokines, and inflammatory mediators are induced by IL-17 [63]. IL-2 is required for the induction of T-cell proliferation. IL-7 and IL-15 drive proliferation of T cells in an antigen-independent manner in lymphopenic conditions [64, 65]. IL-2 suppresses the formation of Th17 cells [66]. IL-4, while inducing Th2 cells, suppresses the formation of induced FoxP3+ T cells and Th1 cells [67, 68]. IL-27 promotes the generation of Tr1 cells [69, 70]. Expression or activation of specific transcription factors is required for the generation of specialized effector T cells and Tregs. For example, RORγt, STAT3, and AHR are important for Th17 cells. FoxP3 and STAT5 are important for the formation of induced Tregs. c-Maf and aryl hydrocarbon receptor (AHR) are important for formation of Tr1 cells [61, 60, 71]. Beyond cytokines, many other factors can modulate the generation of Tregs and Th17 cells. This subject will not be discussed in detail, as the generation of Tregs and Th17 cells during basic immune responses is exhaustively discussed elsewhere.
6.4 Impact of Tumor-Derived Factors on Regulation of T-Cell Differentiation
Most T cells in tumors are memory T cells [72]. Both antigen-specific and nonspecific bystander T cells would be present in tumors. In general, the presence of memory T cells and CD8+ T cells is linked to positive prognosis in cancer patients. This indicates that it is beneficial to have these T cells in tumors. About 30–50 % of CD4+ T cells in various tumors formed in animals are FoxP3+ T cells [72]. Th17 cells are also found in tumors, particularly tumors formed in mucosal tissues [73, 7, 74]. In contrast, Th17 cells are hard to find in transplanted tumors in animal models at ectopic sites [72]. Many factors of the tumor microenvironment can promote the generation of FoxP3+ T cells. First, APCs in tumor environments are prone to generate FoxP3+ T cells. During infection, DCs uptake antigens and undergo maturation in response to TLR activation. Activated DCs emigrate tissue sites of infection and migrate into secondary lymphoid tissues through lymphatic vessels. Only mature DCs express MHC molecules and co-stimulatory molecules such as B7-1 and B7-2 at high levels. In tumors, the signals to maturate DCs are diverse and not as apparent as those in infection. Thus, APCs maturated in tumor microenvironment do not highly express the co-stimulatory molecules [75]. Moreover, tumor-associated APCs express co-inhibitory receptor ligands such as PD-L1 and PD-L2 [76, 77]. This affects T-cell activation and differentiation. Therefore, DCs in or from tumors have low activation potentials for T cells. This condition typically generates induced FoxP3+ T cells but not effector T cells. Other APCs in tumors, such as macrophages and MDSC, are also ineffective in generating effector T cells but are prone to induce Tregs [78].
As mentioned, the hypoxic condition in the tumors is another regulatory factor for T cells [79]. It is expected that draining lymph nodes or tertiary lymphoid tissues within tumors have low oxygen levels. T cells become FoxP3+ T cells when they are activated in hypoxia [80]. This is in part mediated by a transcription factor called HIF-1α. The high glycolytic activity in tumors leads to accumulation of lactic acid [81–83]. This promotes the generation of FoxP3+ T cells. TGF-β1 is a characteristic cytokine produced in the tumor environment [84–86]. TGF-β1 is the most efficient cytokine that induces FoxP3+ T cells in the periphery. Along with TGF-β1, IL-10 acts to suppress antitumor immune responses and the promotion of Tregs [87, 88]. IL-10 is produced by various cell types, including T cells, myeloid cells, B cells, and tumor cells.
PGE2 is highly produced in the tumor environment. PGE2 induces FoxP3+ T cells. This induction is mediated by EP4 and EP2 receptors [89, 90]. In this regard, inhibition of cyclooxygenase-2 (COX-2) decreased FoxP3 expression in tumors and reduced tumor burden [91]. Interestingly, FoxP3+ Tregs express COX-2 and produce PGE2 [92]. The PGE2 produced by Tregs suppresses effector T cells. In addition, prostaglandin D2 (PGD2) acts on DCs to induce FoxP3+ T cells [93]. This effect is mediated through the D prostanoid receptor and cyclic AMP-dependent protein kinase A. In this regard, enforced expression of COX-2 in head and neck squamous cell carcinoma led to expansion of IL-10+ FoxP3+ T cells [94].
Commensal bacterial products that activate TLR2 are implicated in selectively promoting FoxP3+ T cells and Th17 cells. Segmented filamentous bacteria (SFB) promote Th17 cells in the small intestine [95]. Certain bacterial groups such as Clostridium and Bacteroides fragilis promote the generation of FoxP3+ T cells in the intestine [96, 97]. Tumors, formed in the intestine, female reproductive tract, and skin, are expected to be heavily influenced by commensal bacteria. In these tumors, bacterial MAMPs would activate APC and T cells to regulate the generation of FoxP3+ T cells and Th17 cells. Thus, depending on the bacterial group that is dominant in the tumor environment, FoxP3+ T cells and Th17 cells can be differentially generated.
As mentioned, retinoic acid is an important tumor factor. Retinoic acid affects T cells and tumor cells. Retinoic acid promotes the generation of FoxP3+ T cells but suppresses that of Th17 cells [98, 99]. Retinoic acid affects the development of DCs and generates tolerogenic DCs expressing Arg1 [100]. These DCs promote the generation of FoxP3+ T cells but suppress the formation of Th17 cells. This function seems to be mediated through RAR-α. It is also reported that retinoic acid at low concentrations (i.e., 0.5–5 nM) is required for normal function of effector T cells [101, 102]. Low concentrations of RA are found in bodily fluids in most tissues. In vitamin A deficiency, the migration and function of effector T cells are severely impaired. As mentioned, tumor cells express CYP26 and can decrease retinoic acid concentration in tumors and associated tissues [58]. This hyporetinoic acid condition would significantly affect the T-cell profile in tumors and associated lymphoid tissues. Moreover, retinoic acid can promote differentiation of tumor-associated MDSC into dendritic cells and macrophages [103].
6.5 Migration of Tregs and Th17 Cells into Tumors
Migration of T cells, including Tregs and Th17 cells, is regulated by trafficking receptors such as chemokine receptors and adhesion molecules [104, 105]. Adhesion molecules such as selectins and integrins mediate rolling and firm adhesion of leukocytes on endothelial cell vessels [106, 107]. Chemokines induce integrin activation between rolling and firm adhesion of immune cells on endothelial cells. Chemokines also induce chemotaxis for migration of immune cells within tissues. Organs and tissues express distinct and overlapping chemokines and adhesion molecules for regulation of immune cell migration [108]. Since tumors are formed within specialized organs and tissues, there are similarities in expression of trafficking signals between normal tissues and tumors formed within the tissues. Compared to normal tissues, however, tumors have altered expression of chemokines and adhesion molecules [109]. The trafficking signals and receptors required for T-cell migration into the intestine are well established. In the intestine, CCL20 and CCL25 are, respectively, expressed in the subepithelial cell dome (SED) of Peyer’s patches and by small intestinal epithelial cells [110–113]. Endothelial cells in the intestine, Peyer’s patches, and mesenteric lymph nodes express mucosal addressin cell adhesion molecule-1 (MAdCAM-1) [114]. T cells migrating to the small intestine express CCR9 and α4β7 [115–117]. Memory T cells migrating to the Peyer’s patches express CCR6 [118, 119]. Naïve T cells migrating to Peyer’s patches, MLN, and PLN express CCR7, α4β7, and CD62L [120]. Memory T cells migrating to other tissues or inflamed tissues variably express CCR1-6, CCR8, CCR9, CCR10, CXCR3, CXCR5, and CXCR6 [108]. Effector T cells frequently express P-selectin glycoprotein ligand-1 (PSGL-1), E-selectin ligand-1 (ESL-1), CXCR3, CCR5, and CCR4 [105, 120].
The trafficking receptors of Tregs and Th17 cells have been determined. FoxP3+ T cells that are freshly made in the thymus express CCR7, CXCR4, and CD62L [121, 122]. FoxP3+ T cells activated or induced in the periphery express memory-type trafficking receptors that are frequently expressed by Th1 or Th2 cells. Th17 cells express most memory-type chemokine receptors [123, 124]. CCR6 is a characteristic chemokine receptor expressed by most Th17 cells. In general, FoxP3+ Tregs and Th17 cells follow the trafficking pattern of conventional naïve and memory/effector T cells. Conventional naïve CD4+ T cells expressing CCR7 and CD62L lose these receptors upon T-cell activation in the secondary lymphoid tissues and migrate into nonlymphoid or inflamed tissues. Various tissue factors influence the expression of trafficking receptors on FoxP3+ T cells and Th17 cells [125, 126]. For example, retinoic acid acts on T cells undergoing activation to induce gut-homing receptors such as CCR9 and α4β7. FoxP3+ T cells and Th17 cells express these gut-homing receptors and migrate to the intestine [98, 127]. In vitamin A deficiency, the number of FoxP3+ T cells and Th17 cells in the gut is significantly decreased in part because most T cells do not migrate to the small intestine [128]. In addition, TGF-β1 is a major cytokine that induces the expression of CCR6 on FoxP3+ T cells and Th17 cells [123]. Moreover, IL-2 is a cytokine that effectively downregulates CCR6 expression induced by TGF- β1. Thus, cytokines and tissue factors can co-regulate the expression of trafficking receptors on T cells.
Researchers have been searching for chemokines that regulate immune cell trafficking and antitumor immune responses [129–133]. Chemokines such as CCL3-5, CCL20, and CXCL10, often expressed in inflamed tissues, are also expressed in tumors [134–139]. Chemokines induce chemotaxis of immune cells and tumor cells. They can co-stimulate T cells and promote angiogenesis [140, 141]. CCR2-10 and CXCR3-5 regulate T-cell trafficking in various tumors [132]. Most of these receptors are highly expressed by FoxP3+ T cells and Th17 cells in mice and humans [105, 123, 124, 121, 122, 142]. CCL17 and CCL22 are highly expressed in gastric cancer with CCR4-expressing FoxP3+ T cells [131]. CCR7 is expressed by some T cells in colorectal cancers and is predictive of positive prognosis [143]. CXCR4+ T cells are increased in lung adenocarcinoma [144]. Chemokines expressed in tumors also attract hematopoietic progenitors, myeloid cells, NK cells, and CD8+ T cells [136, 145, 10]. An important point is that chemokine signals in cancer patients are highly diverse among different tumors. They are also affected by tissue sites and inflammatory responses in tumors. Therefore, it is difficult to find universal trafficking signals which govern T-cell trafficking in most tumors.
Our group investigated the trafficking receptors expressed by tumor-infiltrating FoxP3+ T cells [72]. FoxP3+ T cells account for 25–50 % of CD4+ T cells infiltrating A20, CT26, 4T1, and B16 tumors. Most of these FoxP3+ T cells are memory CD44+ CD62– T cells, which are downregulated for CD62L and CCR7. Downregulation of CCR7 was critical for the migration of FoxP3+ T cells into tumors, as CCR7high FoxP3+ T cells were not efficient at migrating into tumors [72]. Downregulation of CCR7 and CD62L occurs in tumor-draining lymph nodes during antigen priming. Therefore, migration of T cells into secondary lymphoid tissues is required to acquire a proper trafficking receptor phenotype for migration into tumors. While downregulated for CCR7 and CD62L, tumor-infiltrating FoxP3+ T cells express CCR8 and CXCR4 at high levels [72]. This trafficking receptor phenotype reflects the differentiation status of the tumor-infiltrating T cells and/or the trafficking receptor requirement for FoxP3+ T-cell migration into the tumors. Induction of FoxP3+ T cells from FoxP3– T cells in tumors was assessed, and the results indicate that this induction is inefficient [72]. Thus, the tumor-infiltrating FoxP3+ T cell in these tumors is largely from the FoxP3+ T cells made in the thymus or secondary lymphoid tissues rather than FoxP3+ T cells induced directly in tumors. However, this can be quite different in other types of tumors where the tumor microenvironment is more conducive in priming T cells for differentiation into Tregs. In tumors, FoxP3+ T cells appear highly stable in maintaining their FoxP3 expression. While detailed information on Th17 cell migration into tumors is not available, Th17 cells would probably utilize the same tissue- or inflammation-associated trafficking signals utilized by Th17 cells for regulation of general immune responses. Th17 cells are prevalent in the gastrointestinal (GI) tract and other mucosal tissues. High numbers of Th17 cells were observed in aggressive forms of GI cancers [73, 7, 74]. Thus, these tumors would have trafficking and cytokine signals appropriate for recruitment and maintenance of Th17 cells or their progenitors. Migration of FoxP3+ T cells and Th17 cells into tumors and draining lymph nodes is summarized in Fig. 6.2.
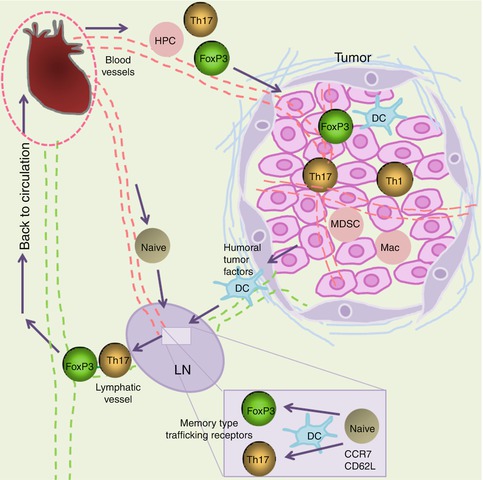
Fig. 6.2
Migration of FoxP3+ T cells and Th17 cells into tumors. Natural FoxP3+ T cells made in the thymus can migrate into lymph nodes, but cannot migrate directly into tumors unless tumors are formed in lymphoid tissues. FoxP3+ T cells can migrate into tumors after they are antigen primed in secondary lymphoid tissues and gain the memory/effector-type trafficking receptors. Loss of CCR7 and CD62L occurs during antigen priming and is required for migration of antigen-primed FoxP3+ T cells into tumors. Induced FoxP3+ T cells in the tumor-draining lymph nodes can migrate into tumors, as they are downregulated for CCR7 and CD62L but upregulated for memory/effector-type trafficking receptors such as CCR4, CCR5, CCR8, CCR10, and/or CXCR4. Dendritic cells (DCs) transport and present tumor tissue antigens and play important roles in the generation of FoxP3+ T cells and Th17 cells in lymph nodes. Soluble tumor tissue factors are collected in tumor-draining lymph nodes, and some affect T-cell priming and differentiation. In tumors, macrophages (Mac), DCs, and MDSC suboptimally activate T cells in tumors. These APCs play potentially important roles in maintaining the phenotype of FoxP3+ T cells and Th17 cells in tumors. There is no such thing as tumor-specific trafficking receptors. Instead, T cells variably use conventional trafficking receptors to migrate into different tumors
6.6 Impact of Tregs and Th17 Cells on Antitumor Immune Responses
The presence of T cells in tumors is a highly reliable prognostic factor for survival of cancer patients [146, 147]. There is a strong positive correlation between patient survival and frequencies of memory CD4+ T cells and CD8+ T cells in many cancer types. Tumorigenesis is increased in pan-T-cell- or γδ-T-cell-deficient animals or humans [148]. Strikingly, αβ T cells have a small negative effect on tumor numbers, but a greater positive effect on tumor size. This implies that αβ T cells are composed of heterogeneous subsets with different functions, and some of these T cells may even promote tumor growth. FoxP3+ T cells and other regulatory T cells are likely the T cells that suppress antitumor immune responses. FoxP3+ T cells can inhibit antitumor immune responses and promote tumor growth [149]. Many FoxP3+ T cells are self-reactive and effective in preventing autoimmune diseases. The same function can be used to promote tumor growth. This is because tumor cells basically express self-antigens, and FoxP3+ T cells can effectively suppress immune responses to self-antigens [150]. In the same line, the frequencies of FoxP3+ T cells in many tumor types are inversely correlated with patient survival rates [151, 147]. However, lack of correlation or positive correlation has been noticed as well [152, 153]. A good example is colorectal carcinoma, in which high frequencies of FoxP3+ T cells are associated with a favorable prognosis [5]. It is expected that FoxP3+ T cells can even prevent the formation of some tumors by suppressing tissue inflammation at early stages of tumorigenesis. Therefore, FoxP3+ T cells have the potential to either promote or suppress tumorigenesis depending on tumor type, tissue site, and immune response. The potentially complex functions of Tregs in tumorigenesis are depicted in Fig. 6.1.
It has been observed that Th17 cells can promote CD8+ T-cell-mediated antitumor immune responses in a mouse model [154]. Moreover, polarization of CD8+ T cells into Tc17 cells increased their antitumor immunity [155]. Th17 cells may become Th1 cells or activate CD8+ T cells to increase antitumor immunity. Paradoxically, Th17 cells can cause inflammation to initiate development of inflammatory tumors at early stages of tumorigenesis. In colorectal cancer, Th17 cells are linked to poor prognosis, whereas Th1 cells are positively linked to patient survival [156]. The major cytokine product of Th17 cells, IL-17, can induce tissue inflammation and the expression of certain angiogenic factors, including CXCL8, MMP-2, MMP-9, and VEGF [157]. The function of Th17 cells in cancer can be complex and appears to be determined again by cancer type, stage, and site. The potentially complex functions of Th17 cells in tumorigenesis are depicted in Fig. 6.1.
Apart from their effector functions, the frequencies of FoxP3+ T cells and Th17 cells reflect the context of the tumor microenvironment. Noninflammatory tumors with low expression of IL-6 and other inflammatory cytokines would have high numbers of FoxP3+ T cells, whereas inflammatory tumors with high expression of inflammatory cytokines would harbor high numbers of Th17 cells. Tumors are heterogeneous in the tumor microenvironment even within the same group of cancers, and not all tumors fit into the inflammatory vs. noninflammatory tumor model. While there is an inverse correlation between FoxP3+ T cells and Th17 cells, both T-cell subsets can be increased or decreased depending on the balance of cytokines and other tissue factors. An example for this situation is invasive ductal breast carcinoma [157].
6.7 Concluding Remarks
As discussed throughout this chapter, FoxP3+ T cells and Th17 cells play both positive and negative roles in regulating antitumor immune responses (Fig. 6.1). Despite the presence of these T cells, some tumors still develop and grow. Thus, these T cells by themselves are not sufficient to effectively mount antitumor immune responses. More detailed studies on FoxP3+ T cells and Th17 cells in various tumors can provide systematic information regarding the tumor microenvironment and therapeutic interventions. It is important to develop novel strategies to boost the beneficial effects of the T-cell subsets and to suppress their tumor-promoting effects. The key is to alter tumor microenvironment to regulate these T-cell subsets. This is expected to be achieved through control of antigen-presenting cells, metabolism, cytokines, chemokines, co-stimulatory/inhibitory receptors, inflammatory mediators, and nuclear hormone receptor ligands such as retinoic acid. Regulation of multiple factors at the same time would provide more effective strategies in tipping the T-cell balance toward tumor-eradicating immune responses. A one-size-fits-all approach is not likely to be effective in changing the microenvironment and T-cell activity in all tumors. In this regard, another point is that antitumor therapy strategies should be tailor-made based on cancer type, tissue site, and tumor microenvironment. It is expected that application of wrong immunotherapy strategies to regulate the T-cell subsets could even worsen the prognosis of cancer patients. More research into classification of cancer types based on tumor microenvironment and immunological milieu would be highly useful.
Acknowledgments
The author thanks Kim Lab members and F. Chu (Purdue University) for their inputs and assistance in preparation of this chapter. This study was supported, in part, by grants from the NIH (R01AI074745, R01DK076616, 1R01AI080769, and 1S10RR028293), the Crohn’s and Colitis Foundation of America, and the National Multiple Sclerosis Society to CHK.
References
1.
Rolle CE, Sengupta S, Lesniak MS. Mechanisms of immune evasion by gliomas. Adv Exp Med Biol. 2012;746:53–76.PubMed
2.
Morse MA, Hall JR, Plate JM. Countering tumor-induced immunosuppression during immunotherapy for pancreatic cancer. Expert Opin Biol Ther. 2009;9(3):331–9.PubMed
3.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.PubMed
4.
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88.PubMed
5.
Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother. 2011;60(7):909–18.PubMed
6.
Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol. 2011;186(7):4388–95.PubMed
7.
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114(6):1141–9.PubMedCentralPubMed
8.
Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res. 2008;14(11):3254–61.PubMedCentralPubMed
Related posts:
 Prognostic Value of Innate and Adaptive Immunity in Cancers
Prognostic Value of Innate and Adaptive Immunity in Cancers
 Autophagy and Necroptosis in Cancer
Autophagy and Necroptosis in Cancer
 MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
MHC Class I Molecules and Cancer Progression: Lessons Learned from Preclinical Mouse Models
 B Cells in Cancer Immunology: For or Against Cancer Growth?
B Cells in Cancer Immunology: For or Against Cancer Growth?
 Immunohistochemistry of Cancers
Immunohistochemistry of Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


