Receptor transduction pathways mediating hormone action
CLASS A RECEPTORS THAT TRANSDUCE HORMONE ACTION
The Glycoprotein Hormone Receptor Group
The Gonadotropin–Releasing Hormone Receptor Group
The Thyrotropin-Releasing Hormone and Secretagogue Receptor Group
CLASS B RECEPTORS THAT TRANSDUCE HORMONE ACTION
Growth Hormone–Releasing Hormone Receptor
Gastric Inhibitory Polypeptide Receptors
Parathyroid Hormone and Parathyroid Hormone–Related Peptide Receptors
CLASS C RECEPTORS THAT TRANSDUCE HORMONE ACTION
THE INSULIN-LIKE GROWTH FACTOR-1 RECEPTOR
THE FIBROBLAST GROWTH FACTOR RECEPTOR FAMILY
SUBFAMILY 1 NUCLEAR RECEPTORS: THYROID HORMONE, VITAMIN D3, AND PEROXISOME PROLIFERATOR-ACTIVATED RECEPTORS
SUBFAMILY 2 NUCLEAR RECEPTORS: HEPATOCYTE NUCLEAR FACTOR AND RETINOID X RECEPTORS
SUBFAMILY 3 NUCLEAR RECEPTORS: THE STEROID RECEPTORS AND GLUCOCORTICOID, ANDROGEN, ESTROGEN, AND MINERALOCORTICOID RECEPTORS
SUBFAMILY 0 NUCLEAR RECEPTORS: DAX1
Introduction
Hormones exert their actions by binding to specific receptor proteins, a process that induces conformational changes or compartmental redistribution of these proteins. The activated receptor is now capable of inducing positive (or negative) intracellular effects that ultimately are recognized as a physiologic response. The specificity of hormone action is determined by the affinity of hormones for different receptors, the cell-specific expression of the receptor, and the unique responses induced by ligand occupancy.
Since the early 2000s, our understanding of hormone action has advanced rapidly with the success of genomics and advanced molecular biologic techniques. This combined approach has led to the discovery and classification of an unexpectedly large number of receptors, some quite novel and others even unanticipated, that are members of large families of genetically conserved proteins. Moreover, our understanding of receptor action has been clarified by the identification and detailed characterization of postreceptor signaling proteins and signaling mechanisms. Four major receptor superfamilies have been identified that are distinguished by protein structure, cellular localization, and effector systems. These families include the G protein–coupled receptors (GPCRs), cytokine receptors, tyrosine kinase receptors (RTKs), and nuclear receptors (Table 3-1). This chapter reviews major features of these important receptor families. Mutations influencing receptor function leading to endocrine disorders are also highlighted.

G protein–coupled receptors
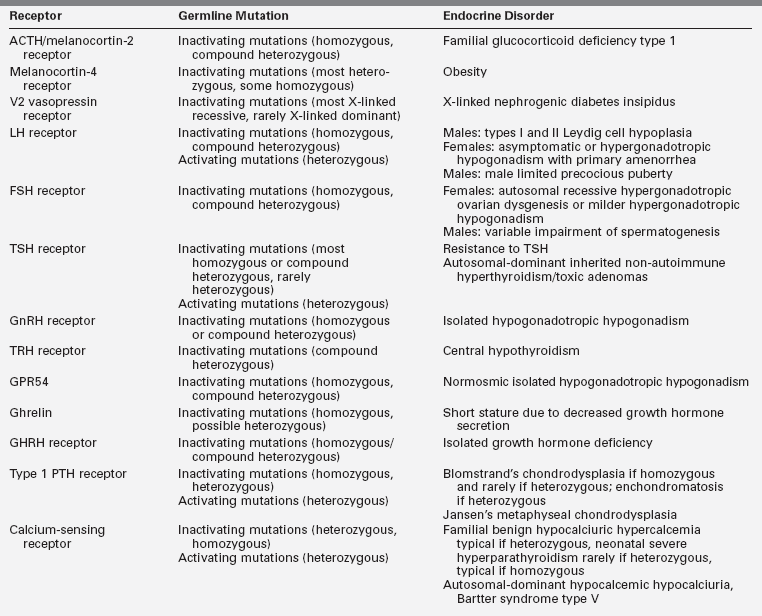
More than 1% of the genome of vertebrates encodes a large protein family of receptors that sense molecules outside the cell and activate inside signal transduction pathways and, ultimately, cellular responses. These receptor proteins are embedded in the plasma membrane and are coupled to intracellular signal generating systems by heterotrimeric G proteins (i.e., G protein coupled receptors [GPCRs]).1 GPCRs are also known as seven-transmembrane domain receptors, 7TM receptors, heptahelical receptors, and serpentine receptors. They are called transmembrane receptors because they pass through the cell membrane, and they are called seven-transmembrane receptors because they pass through the cell membrane seven times. The human genome encodes roughly 950 G protein–coupled receptors, which detect photons (light), hormones, growth factors, drugs, and other endogenous ligands. Approximately 150 of the GPCRs found in the human genome have unknown functions. Most GPCRs are odorant and pheromone receptors.1 Also important to note is that most hormones bind to GPCRs, and hence G protein–dependent signal transduction represents the most common mechanism for hormone action (Table 3-2).
G protein–coupled receptors are involved in many diseases and are also the target of approximately 40% of all modern medicinal drugs.2 G proteins were discovered when Alfred G. Gilman and Martin Rodbell investigated stimulation of cells by adrenaline. These investigators discovered that when adrenaline binds to a receptor, the receptor does not stimulate enzymes directly. Instead, the receptor stimulates a GTP binding protein, which stimulates an enzyme. An example is adenylate cyclase (AC), which produces the second messenger cyclic AMP. For this discovery, they won the 1994 Nobel Prize in physiology or medicine. The 2012 Nobel Prize in chemistry was awarded to Brian Kobilka and Robert Lefkowitz for their work that was “crucial for understanding how G protein–coupled receptors function.”
The GPCR superfamily is divided into eight major classes.1,3 These receptors contain an N-terminal extracellular domain that is frequently called the ectodomain or exodomain.4 These receptors also contain seven putative transmembrane spanning alpha helices (TM-I to TM-VII). The alpha helices are connected by three intracellular (i1 through i3) and three extracellular (e1 through e3) loops that are often collectively called the serpentine region (Figure 3-1).4,5 The C-terminal intracellular region is usually referred to as the endodomain.4
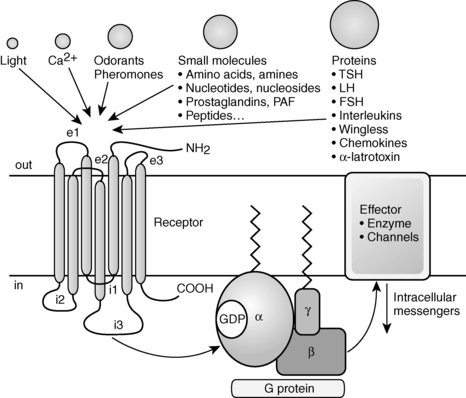
FIGURE 3-1  GPCR structure and function. GPCRs have an N-terminal extracellular domain, seven putative transmembrane domains separated by three extracellular loops (e1-e3) and three intracellular loops (i1-i3), and a C-terminal intracellular domain. Ligand binding results in the exchange of GTP for GDP, which induces dissociation of the G protein into a GTPα subunit and a βγ subunit. Then these subunits alter the activity of intracellular effector enzymes and transmembrane channels, resulting in the alteration of intracellular levels of second messengers that can include cAMP and calcium. (Adapted with permission from Bockaert, J., & Pin, J. P. (1999). Molecular tinkering of G-protein–coupled receptors: an evolutionary success. EMBO J, 18, 1724.)
GPCR structure and function. GPCRs have an N-terminal extracellular domain, seven putative transmembrane domains separated by three extracellular loops (e1-e3) and three intracellular loops (i1-i3), and a C-terminal intracellular domain. Ligand binding results in the exchange of GTP for GDP, which induces dissociation of the G protein into a GTPα subunit and a βγ subunit. Then these subunits alter the activity of intracellular effector enzymes and transmembrane channels, resulting in the alteration of intracellular levels of second messengers that can include cAMP and calcium. (Adapted with permission from Bockaert, J., & Pin, J. P. (1999). Molecular tinkering of G-protein–coupled receptors: an evolutionary success. EMBO J, 18, 1724.)
GPCRs are activated by a wide variety of signals, including proteins, nucleotides, amino acid residues, Ca2+, light photons, and odorants (see Figure 3-1).1 It is postulated that ligand binding alters the conformation of transmembrane domains and intracellular loops, increasing the affinity of the receptor for specific heterotrimeric guanosine nucleotide binding proteins (G proteins) (see Figure 3-1).6,7 G proteins share a common heterotrimeric structure consisting of an α subunit and a tightly coupled βγ dimer. The α subunit interacts with detector and effector molecules, binds guanosine 5’-triphosphate (GTP), and possesses intrinsic GTPase activity. There are 16 genes in mammals that encode some 20 different α chains. The Gα subunits are categorized in four classes and include Gsα (G stimulatory), Giα (G inhibitory) and Goα (G other), Gq/11α, and G12/13α. They behave differently in the recognition of the effector but share similar structures and mechanism of activation. The Gα subunits consist of two domains: a GTP-binding domain and a helical insertion domain. The GTP-binding domain is homologous to Ras-like small GTPases and includes switch regions I and II, which change conformation during activation. The switch regions are loops of alpha helices with conformations sensitive to guanine nucleotides. The helical insertion domain is inserted into the GTP-binding domain before switch region I and is unique to heterotrimeric G proteins. This helical insertion domain sequesters the guanine nucleotide at the interface with the GTP-binding domain and must be displaced to enable nucleotide dissociation.
The α subunits associate with a smaller group of β(5) and γ(12) subunits.8 Combinatorial specificity in the associations between various G protein subunits provides the potential for enormous diversity and may allow distinct heterotrimers to interact selectively with only a limited number of G protein–coupled receptors and effector proteins.6,7,9
G protein signaling is terminated by the hydrolysis of α-GTP to α-GDP by an intrinsic GTPase. A group of proteins, called regulator of G protein signaling (RGSs), acts as GTPase-activating proteins (GAPs), specific for Gα subunits. These proteins accelerate hydrolysis of GTP to GDP and terminate the transduced signal. In some cases, the effector itself may possess intrinsic GAP activity, which helps deactivate the pathway. This is true in the case of phospholipase C β, which possesses GAP activity within its C-terminal region. This is an alternate form of regulation for the Gα subunit. However, it should be noted that the GAPs do not have catalytic residues to activate the Gα protein. Rather, GAPs reduce the required activation energy for the reaction to take place. After hydrolysis of GTP, the Gα-GDP chain reassociates with the βγ dimer; the reassociated heterotrimeric G protein is now capable of participating in another cycle of receptor-activated signaling.1,6,7,9
Specificity in ligand binding is conferred by variations in the primary structures of the extracellular and intracellular domains.1 Specificity of effector responses is conferred by the variations in the primary structure of intracellular domains and isoforms of the Gα subunits of G proteins.10,11 Some GPCRs couple predominantly with Gαi /Gαo subunits that act primarily to decrease adenylyl cyclase activity.11–14 Other GPCRs couple predominantly with Gαs subunits that increase adenylyl cyclase activity or Gαq/Gα13,16–18 subunits that increase phospholipase C activity.11,15,16
Interestingly, data show that cytoskeletal proteins may modulate receptor–G protein coupling. For example, the erythrocyte membrane cytoskeletal protein 4.1G can interfere with A1 adenosine receptor signal transduction.17 4.1G also influences metabotropic glutamate receptor 1α-mediated cAMP accumulation, increases the ligand-binding ability of metabotropic glutamate receptor 1α, and alters its cellular distribution.18 4.1G may also play a role in receptor-receptor dimerization.
Receptor agonist-independent and agonist-induced homo- and heterodimerization have increasingly been recognized as important determinants of GPCR function.19 For example, the GPCR somatostatin receptor 5 (SSTR5) primarily exist as monomers in the absence of an agonist. However, they form homodimers in the presence of an agonist.20 Furthermore, it has been shown that SSTR5 can form heterodimers with type 2 dopamine receptors (DRD2)—another GPCR—in the presence of hsst2 agonist or dopamine.21 Agonist-induced activation of SSTR5-DRD2 heterodimers in Chinese hamster ovary (CHO) cells expressing SSTR5 and DRD2 is increased when compared to agonist-induced activation of monomers and homodimers in CHO cells expressing only SSTR5 or DRD2.21 Heterodimerization of receptors may also lead to inactivation of one of the receptors in the complex. For example, heterodimerization of somatostatin receptor 2A (sst2A) with somatostatin receptor 3 (SSTR3) appears to lead to inactivation of the heterodimerized SSTR3 without inactivating the heterodimerized SSTR2.22
GPCRs can form heterodimers with nonreceptor transmembrane proteins. Both the calcitonin receptor (CALCR) and the calcitonin receptor-like protein (CALCRL) can form heterodimers with three different accessory proteins that are termed “receptor-activity–modifying proteins (RAMPs)”: RAMP1, RAMP2, and RAMP3.23–25 Whereas CALCRs can be activated by ligand in the absence of heterodimerization with a RAMP, CALCRLs are only activated by ligand if heterodimerized with a RAMP.23,24 RAMPs alter the ligand specificity of the heterodimerized receptor.
CALCRs that are not in heterodimers with RAMPS are activated by calcitonin and thus constitute the classic CALCR.23,24 However, CALCRs heterodimerized with RAMP1, RAMP2, and RAMP3 bind amylin and constitute amylin1, amylin2, and amylin3 receptors, respectively.23,24 CALCRLs dimerized with RAMP1 bind calcitonin gene–related peptide and constitute the calcitonin gene–related peptide receptor.23,24 CALCRLs dimerized with RAMP2 and RAMP3 bind adrenomedullin and constitute adrenomedullin1 and adrenomedullin2 receptors, respectively.23,24 RAMPS alter function of other GPCRs that transduce hormone action. The distribution and function of parathyroid hormone 1 and 2 receptors are altered by binding to RAMP2 and RAMP3, respectively.26 The distribution and function of the glucagon receptor is altered by binding to RAMP2.26 Dimerization/heterodimerization may occur in the endoplasmic reticulum (ER) shortly after protein synthesis occurs.27 The ER plays a role in determining whether or not a protein will be expressed elsewhere in the cell, thus protecting the cell from misfolded and (likely) mutant proteins.27 The non-heterodimerized CALCRL is an orphan receptor because the CALCRLs cannot leave the ER for the cell membrane unless heterodimerized with RAMPs.28
The melanocortin receptors also utilize accessory proteins. Circulating ACTH binds to five different forms of the melanocortin receptor (types 1-5), but only the melanocortin 2 receptor (MC2R) in the adrenal cortex leads to release of adrenal steroids. MC2R interacts with Gs, which leads to activation of adenylyl cyclase and formation of cAMP. The MC2R is the smallest G protein–coupled receptor known to date and belongs to a family of melanocortin receptors (types 1 to 5) that bind to various derivatives of proopiomelanocortin, especially α-MSH. The accessory protein melanocortin 2 receptor-associated protein (MRAP) is required for MC2R function, as it is critical for the translocation of the receptor from the endoplasmatic reticulum to the cell surface.29 Moreover, MRAP facilitates signaling of the MC2R.30 Loss of function of MRAP thus prevents membrane expression of MC2R and completely prevents ACTH signaling. Interestingly, MRAP forms a unique antiparallel homodimer in close proximity to the MC2R.31 The MRAP accessory protein can also interact with other melanocortin receptors, particularly MC5R, but exerts negative effects on their signaling. Expression of MRAP was shown to be predominantly present in the zona fasciculata in the rat adrenal gland, consistent with its facilitating role in glucocorticoid production. Hence, mutations in MC2R32 or MRAP29 can lead to familial glucocorticoid deficiency secondary to ACTH resistance. By contrast, MRAP2, a protein with 39% amino acid homology to MRAP, shares the MC2R-trafficking function of MRAP but does not appear to play a major supportive role in adrenocortical ACTH signaling. On the contrary, in vitro studies have shown that overexpression of MRAP2 can suppress MC2R activation.
Failure of the endoplasmic reticulum to export mutant GPCR homodimers and mutant GPCR wild-type GPCR heterodimers to the cell membrane has been found to be the cause of dominant negative endocrine conditions. A dominant negative mutation is a heterozygous mutation that results in a phenotype that would only be expected in the presence of a homozygous mutation. Some heterozygous MC4R mutations cause dominantly inherited obesity due interaction of wild-type MC4R with the mutant receptor, and this specific effect of protein-protein interaction results in a dominant-negative effect.33,34 In addition, some V2 vasopressin receptor gene mutations that are known to cause nephrogenic diabetes insipidus encode mutant V2 vasopressin receptors that form dimers in the ER that cannot be exported to the cell membrane.35
These mutant receptors also interfere with cell surface expression of wild-type receptors by forming heterodimers with the wild-type receptors that cannot be exported from the ER to the cell membrane.36 This finding explains why females heterozygous for these V2 vasopressin receptor gene mutations do not concentrate their urine with even high doses of desmopressin, a synthetic V2 vasopressin receptor agonist, in spite of being able to produce wild-type V2 vasopressin receptors.37 A similar phenomenon explains dominant transmission of partial TSH receptor resistance in patients heterozygous for some inactivating TSH receptor mutations.38 In these patients, mutant TSH receptors form oligomers with wild-type receptors and prevent export of wild-type receptors from the endoplasmic reticulum to the cell membrane.38
Similarly, misfolding and misrouting of some mutant gonadotropin-releasing hormone (GnRH) receptors in the endoplasmic reticulum (as well as oligomerization of these mutant GnRH receptors with wild-type GnRH receptors) decrease cell membrane expression of wild-type GnRH receptors.39–41 This phenomenon, however, has not been found to have clinical implications in relatives of probands homozygous or compound heterozygous for mutations that cause isolated hypogonadotropic hypogonadism (IHH) because individuals heterozygous for these mutations demonstrate an intact GnRH-gonadotropin axis and do not have clinical signs of IHH. Thus, in these individuals enough wild-type GnRH receptors do not oligomerize with mutant GnRH receptors and are transported to the cell membrane to maintain sufficiently normal GnRH-GnRH receptor interactions to avoid development of IHH.40
GPCRs activate G proteins at very low levels in the absence of ligand binding, but in some cases genetic mutations that lead to substitution of a single amino acid can greatly increase the interaction rate of the unliganded receptor for its G protein. Hence, GPCRs that are specific for luteinizing hormone, thyroid-stimulating hormone, thyrotropin-releasing hormone, glucagon-like peptide-1, melanocortin, and cannabinoid receptors can activate G proteins in the absence of ligand binding,42,43 demonstrating constitutive activity that increases linearly with increased cell surface expression of the receptors.44 It has also been recognized that there are ligands (often called inverse agonists) that decrease the activity of these receptors.45 Receptor ligands that neither increase nor decrease the activity of receptors are now frequently referred to as neutral antagonists.43
The term antagonists is applied to these ligands because they block activation and inactivation of receptors by agonists and inverse agonists, respectively.43 The term agonist only refers to receptor ligands that increase receptor activity.43 A scale has been formulated to express the continuity in receptor ligand function—from −1 (representing a full inverse agonist), to 0 (representing a neutral antagonist), to +1 (representing a full agonist).43,45 It is possible that inverse agonists play a role in treating medical conditions caused by GPCR mutations that lead to increased constitutional activation of the receptor.43
Receptor desensitization and resensitization play a role in GPCR activity. Since the early 2000s, mechanisms of GPCR desensitization and resensitization have been elucidated. Three processes for receptor desensitization have been described.46,47 The first receptor desensitization process is rapid uncoupling of the G protein from GPCRs.47 This process occurs within seconds to minutes after initiation of the process and occurs as a result of phosphorylation of GPCRs.47 G protein receptor kinases (GRKs) have been increasingly recognized as playing a major role when this process involves homologous desensitization.46
Homologous or agonist-dependent desensitization occurs after agonist activation of the receptor that is desensitized.47 GRK-mediated phosphorylation of serine and threonine residues in the third intracellular loop, or the C-terminal intracellular domain leads to activation of β-arrestins, which in turn inactivate adenylyl cyclase (Figure 3-2).46–49 Second-messenger–dependent protein kinases also contribute to receptor desensitization when this process involves homologous desensitization, but they also participate in receptor desensitization when desensitization involves heterologous desensitization. Heterologous or agonist-independent desensitization occurs as a result of activation of a different receptor from the one that is desensitized.47
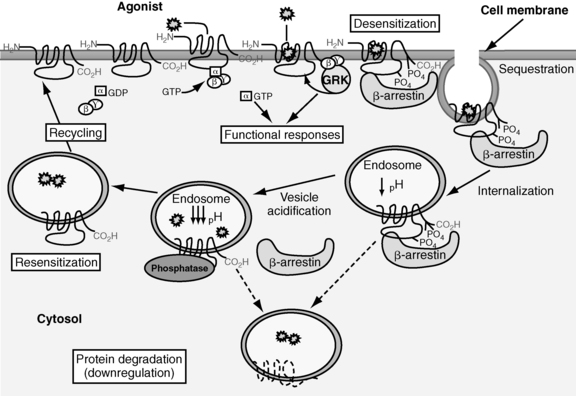
FIGURE 3-2  Desensitization and recycling of GPCRs. Shortly after an agonist binds a GPCR, G-protein–receptor kinases phosphorylation of serine and threonine residues in the third intracellular loop or the C-terminal intracellular domain leads to activation of β-arrestin. Activation of β-arrestin inactivates adenylyl cyclase and initiates sequestration of the GPCR in clathrin-coated vesicles. Dephosphorylation of the sequestered receptor and subsequent disassociation of the receptor from β-arrestin is followed by recycling of the GPCR to the cell membrane. Alternatively, once sequestered the GPCR can be destroyed in lysosomes. (Adapted with permission from Saunders, C., & Limbird, L. E. (1999). Localization and trafficking of α2-adrenergic subtypes in cells and tissues. Pharmacol Ther, 84, 200.)
Desensitization and recycling of GPCRs. Shortly after an agonist binds a GPCR, G-protein–receptor kinases phosphorylation of serine and threonine residues in the third intracellular loop or the C-terminal intracellular domain leads to activation of β-arrestin. Activation of β-arrestin inactivates adenylyl cyclase and initiates sequestration of the GPCR in clathrin-coated vesicles. Dephosphorylation of the sequestered receptor and subsequent disassociation of the receptor from β-arrestin is followed by recycling of the GPCR to the cell membrane. Alternatively, once sequestered the GPCR can be destroyed in lysosomes. (Adapted with permission from Saunders, C., & Limbird, L. E. (1999). Localization and trafficking of α2-adrenergic subtypes in cells and tissues. Pharmacol Ther, 84, 200.)
The second receptor desensitization process is internalization/sequestration of GPCRs. This process is slower than receptor phosphorylation-induced uncoupling of the G protein from GPCRs and occurs within minutes to hours after initiation of the process. This process is reversible because the receptors can be recycled to the cell surface (see Figure 3-2).47 GRKs and β-arrestins play a role in initiating internalization/sequestration of β2-adrenergic, LH, FSH, TSH, TRH, vasopressin V2, angiotensin II type 1A, and other G protein–coupled receptors in clathrin-coated vesicles (see Figure 3-2).46,50–57 Dephosphorylation of the sequestered receptor followed by disassociation of the receptor from β-arrestin is necessary for the receptor to be recycled to the cell membrane and resensitized (see Figure 3-2).16
The third receptor desensitization process is down-regulation. With down-regulation, the number of intracellular GPCRs decreases due to increased lysosomal degradation and decreased synthesis of the receptors due to alteration of transcriptional and posttranscriptional regulatory mechanisms (see Figure 3-2).58,59 Down-regulation is a slow process that occurs within several hours to days after initiation of the processes that lead to its development.60
One of the ways the Arg137His V2 vasopressin receptor mutation interferes with mutant receptor function and causes X-linked nephrogenic diabetes insipidus is by altering desensitization and recycling of the mutant receptor.61 In vitro studies have revealed that the mutant receptor is constitutively phosphorylated. Thus, even in the absence of ligand binding the mutant receptor is bound by β-arrestin—which in turn leads to sequestration of the mutant receptor within clathrin-coated vesicles. Recycling of the mutant receptor back to the cell membrane requires the mutant receptor to be dephosphorylated and disassociated from β-arrestin. However, the mutant receptor remains constitutively phosphorylated while sequestered and thus cannot be disassociated from β-arrestin and recycled to the cell membrane—thereby reducing cell membrane expression of the mutant receptor.
Some investigators suggest that most GPCR-inactivating mutations can be classified by the effects of the mutations into one of five classes.62 Class I inactivating mutations interfere with receptor biosynthesis. Class II inactivating mutations interfere with receptor trafficking to the cell surface. Class III inactivating mutations interfere with ligand binding. Class IV inactivating mutations impede receptor activation. Class V inactivating mutations do not cause discernible defects in receptor biosynthesis, trafficking, ligand binding, or activation but may cause medical disorders. There are also inactivating mutations that interfere with receptor function via multiple mechanisms and thus cannot be placed into one class.
Of the eight classes of GPCRs, only classes A, B, and C contain receptors for mammalian hormones (Figure 3-3).3 Class A receptors contain the rhodopsin-like receptors and are divided into at least 15 groups.3,63 Four of these groups contain receptors activated by hormones. These are the peptide receptor, hormone protein receptor, GnRH receptor, and the thyrotropin-releasing hormone (TRH) and secretagogue receptor groups.3
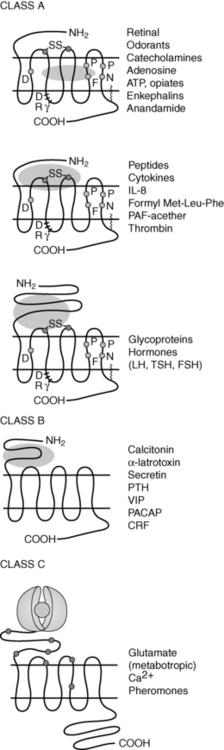
FIGURE 3-3  Examples of class A, B, and C GPCRs. The oval represents the ligand. These receptors can differ in amino acid sequence, in length of the N-terminal extracellular and C-terminal cytoplasmic domains, and in the receptor regions involved with ligand-receptor interactions. (Adapted with permission from Bockaert, J., & Pin, J. P. (1999). Molecular tinkering of G protein-coupled receptors: an evolutionary success. Embo J, 18, 1725.)
Examples of class A, B, and C GPCRs. The oval represents the ligand. These receptors can differ in amino acid sequence, in length of the N-terminal extracellular and C-terminal cytoplasmic domains, and in the receptor regions involved with ligand-receptor interactions. (Adapted with permission from Bockaert, J., & Pin, J. P. (1999). Molecular tinkering of G protein-coupled receptors: an evolutionary success. Embo J, 18, 1725.)
The peptide receptor group includes the angiotensin, adrenocorticotropin hormone (ACTH)/melanocortin, oxytocin, somatostatin, and vasopressin receptors.3 The hormone protein receptor group includes the receptors for glycoprotein hormones, including follicle-stimulating hormone (FSH), luteinizing hormone (LH), and thyrotropin (TSH) receptors.3 These receptors have large extracellular N-terminal domains and ligand-binding sites that include the first and third extracellular loops (see Figure 3-3).1,3 There is also much similarity in amino acid sequence among these receptors (see Figure 3-3).1 The GnRH receptor group only contains the GnRH receptor.2 The TRH and secretagogue receptor group includes the TRH receptor and the growth hormone secretagogue receptor.3
Class B GPCRs are structurally similar to members of the hormone protein receptor group (see Figure 3-3).1 However, unlike the glycoprotein hormone receptors, class B GPCRs do not share similar amino acid sequences.1 This family contains receptors for high-molecular-weight hormones, including calcitonin, glucagon, gastric inhibitory peptide, parathyroid hormone (PTH), and corticotrophin-releasing factor (CRF).1,3,64
Class C receptors have a large extracellular domain with two lobes separated by a hinge region that closes on the ligand (see Figure 3-3).65 This region has also been called the Venus flytrap domain or module due to the trapping mechanism of the hinge region.66 This family includes the calcium-sensing receptor (CASR).1,2
Class A receptors that transduce hormone action
The Peptide Receptor Group
Adrenocorticotropin and Melanocortin-2 Receptors
It is important to note that a newly accepted name for the ACTH receptor is melanocortin-2 receptor (MC2R) because the ACTH receptor is one of five members of the melanocortin receptor family of GPCRs.62 For the purpose of clarity, when discussing interactions between ACTH and its receptor, the older name will be used for the remainder of this chapter. The ACTH receptor gene is located on the short arm of chromosome 18.67 The ACTH receptor has a small extracellular and intracytoplasmic domain. Adrenocorticotropin-induced activation of the ACTH receptor in the zona fasciculata and zona reticularis of the adrenal cortex stimulates Gαs, resulting in increased intracellular cAMP levels that stimulate steroidogenesis by activating cAMP-dependent kinases.68–70
Hereditary isolated glucocorticoid deficiency, resistance to ACTH, and familial glucocorticoid deficiency (FGD) are the same names for an autosomal recessive syndrome that consists of glucocorticoid deficiency accompanied by normal mineralocorticoid secretion. FGD has been classified further as FGD types 1 and 2 and the triple A syndrome.71 Patients with FGD type 1 are homozygous or compound heterozygous for point mutations, resulting in ACTH receptors with abnormal function and account for 25% of FGD cases.71–77 In contrast, patients with FGD type 2 have ACTH resistance due to mutations in melanocotin-2 receptor accessory protein (MRAP).78 Triple A (Allgrove syndrome) is an autosomal recessive syndrome characterized by ACTH-resistant adrenal insufficiency, achalasia, and alacrima—which is caused by mutations in the achalasia-adrenocortical insufficiency-alacrima syndrome (AAAS) gene79 thought to regulate nuclear pore complexes and nucleocytoplasmic transport.80
Patients with FGD type 1 usually present during infancy or early childhood with hypoglycemia.74,82–86 Less commonly, patients may present with a severe infection, frequent minor infections, or childhood asthma that resolves with treatment with physiologic doses of glucocorticoids.71,74,82 Hyperpigmentation thought to be due to increased ACTH levels acting on the MC1R may be seen as early as the first month of life, but usually becomes apparent after the fourth month of life.72–74,82–84,87 There is one reported case of FGD type 1 without hyperpigmentation despite elevated ACTH levels in a patient with homozygous mutations in both the MC2R and the MC1R.88 The assumption that hyperpigmentation is caused by increased ACTH levels acting on the MC1R (both in FGD type 1 and Addison’s disease) was substantiated in this patient whose MC1R mutation had previously been implicated in red hair and pale skin phenotypes. Neonates with FGD type 1 may also suffer from jaundice.74,84,86,89 Tall stature accompanied by an advanced or dissociated bone age, in spite of normal age of onset of puberty, appears to be common in children with FGD type 1.71,74,77,82,83,87 Patients with FGD type 2 have normal heights71,90 and present earlier than patients with FGD type 1. The pathophysiology of tall stature in FGD type 1 is unknown. Both forms of FGD exhibit absent adrenarche confirming the importance of ACTH in the induction and maintenance of adrenarche.78
At presentation, plasma cortisol, androstenedione, and dihydroepiandrosterone levels are low or low normal—and plasma ACTH levels are elevated.71,74,82–86 When supine, patients with FGD type 1 have renin and aldosterone levels that are near normal.71,83,85,86 Histologically, the zona fasciculata and zona reticularis are atrophied with FGD.82 However, demonstrating the lack of an essential role for ACTH in the embryologic development and maintenance of the zona glomerulosa, adrenal cortices in patients with all types of FGD contain zona glomerulosa cells.71,81–83,87,91
Abnormalities in ACTH receptor expression may be seen in other conditions. Evidence suggests that the ACTH receptor-Gαs-adenylyl cyclase-cAMP cascade maintains differentiation of adrenocortical cells and that impairment of this cascade leads to dedifferentiation and increased proliferation of adrenocortical cells.92,93 Adrenocortical carcinomas from some patients have been found to have a loss of heterozygosity (LOH) for the ACTH receptor gene, resulting in markedly decreased ACTH receptor mRNA expression.93 Growth of the tumors with LOH for the ACTH receptor gene also may be more aggressive than the other tumors. An activating mutation of Gi2 that constitutively suppresses adenylyl cyclase activity has also been found in adrenocortical tumors.92 Thus, decreased ACTH receptor activity may be associated with tumorigenesis.
Interestingly, many patients with ACTH-independent macronodular adrenal hyperplasia (AIMAH)—a cause of ACTH-independent Cushing syndrome—exhibit increased glucocorticoid levels in response to noncorticotropin hormones that do not normally induce glucocorticoid release.94–99 These hormones include gastric inhibitory peptide, exogenous arginine and lysine vasopressin, luteinizing hormone, human chorionic gonadotropin, angiotensin II, catecholamines, leptin, and serotonin receptor agonists.94–99 Increased expression of the receptors for these ligands in the abnormal adrenal glands has been implicated as a possible explanation for the abnormal induction of glucocorticoid release by these noncorticotropin ligands.99 However, receptors for some of these ligands are expressed in normal adrenal glands.99 Thus, the mechanism for this phenomenon remains to be fully elucidated.
Other Melanocortin Receptors
Murine studies reveal that melanocortin-3 receptor (MC3R), another of the five members of the melanocortin receptor family, regulates fat deposition as MC3R deficient mice have normal body weight with increased fat mass.62 The role of the MC3R in humans is less clear. More than 24 human MC3R mutations have been identified without evidence of obesity.100,101 However, patients with these mutations were not phenotyped for fat mass to assess whether they phenocopy the mouse model, which exhibits normal body weight with altered partitioning leading to increased fat mass and reduced lean mass.101–103 Homozygosity for a pair of single-nucleotide polymorphisms of the MC3R gene that result in production of partially inactive MC3Rs was found to be associated with pediatric-onset obesity in Caucasian American and African American children.104
The melanocortin-4 receptor (MC4R) is another member of the melanocortin receptor family and plays a role in controlling appetite and weight.105 The MC4R has baseline constitutive (i.e., ligand-independent) activity that can be inhibited by the inverse agonist agouti-related peptide (AgRP).105,106 Activation of the MC4R by its natural agonist α-melanocyte stimulating hormone (α-MSH) produces anorexigenic effects.105,107,108 More than 150 naturally occurring MC4R mutations have been identified, causing hyperphagic obesity, increased lean body mass, increased bone density, and increased linear growth.109–114 Patients with homozygous mutations appear to have more severe obesity than their heterozygous relatives, consistent with codominant inheritance.7,115
MC4R mutations are thought to be the most common monogenic cause of human obesity. The prevalence of pathogenic MC4R mutations in obese populations varies widely ranging from 0.5% to 5.8% depending on the screening criteria and population.116–119 AgRP gene polymorphisms appear to be associated with anorexia nervosa.112,113
Little is known about melanocortin-5 receptors (MC5Rs) in animals and humans. There is only weak evidence from a single linkage and association study of families in Quebec that suggests that MC5Rs may also play a role in regulating body weight and fat mass.114
Another member of the melanocortin receptor family, the melanocortin-1 receptor (MC1R), controls skin and hair pigmentation. Activation of MC1Rs in skin and hair follicle melanocytes by the pro-opiomelanocortin (POMC)-derived peptides α-MSH and ACTH stimulates the synthesis of eumelanin, a brown-black pigment.120,121 Inhibition of MC1R baseline constitutive activity by agouti protein, or specific mutations, leads to release of pheomelanin, a red-yellow pigment, from the melanocytes.121
Inactivating homozygous mutations of the POMC gene cause hypoadrenalism, red hair, fair skin, and early-onset obesity. Hypoadrenalism is characterized by glucocorticoid deficiency due to lack of ACTH production from the POMC precursor. Fair skin and red hair are due to a lack of ACTH and α-MSH-induced melanocyte release of eumelanin that results from activation of MC1Rs. Of note, non-white patients with homozygous POMC mutations do not appear to have the fair skin and red hair phenotype.118,122 In white individuals eumelanin synthesis appears to be dependent on POMC-derived peptides, whereas in darker individuals other genes may control eumelanin synthesis.123 Obesity is due to lack of α-MSH–induced anorectic effects, which normally result when α-MSH activates MC4Rs.124 Heterozygosity for POMC gene mutations has been associated with hyperphagia, early-onset obesity, and increased linear growth.124–126 Both homozygous127,128 and heterozygous129 mutations of prohormone convertase 1 cause obesity in humans. Prohormone convertase 1 acts on POMC, proinsulin, and proglucagon. Patients with prohormone convertase 1 deficiency also have neonatal enteropathy and postprandial hypoglycemia. The cause of enteropathy is unknown but hypothesized to be related to the processing of GLP-2 by prohormone convertase 1. GLP-2 is known to stimulate proliferation and repair of intestinal epithelium.130
Vasopressin Receptors
Nephrogenic diabetes insipidus (NDI) results from decreased responsiveness of the renal tubule to arginine vasopressin (AVP), with resulting excessive loss of free water. NDI is characterized by polydipsia and polyuria that is not responsive to vasopressin and vasopressin analogs.131 Vasopressin binds to the V2 vasopressin receptor (AVPR2), a Gs-coupled receptor, in the basolateral membrane of collecting duct principal cells in the kidney and activates translocation of aquaporin-2 (AQP2) water channels to the apical membrane, thereby inducing water permeability. X-linked NDI is caused by inactivating mutations of the V2 vasopressin receptor (AVPR2) gene located at Xq28 and accounts for about 90% of genetically determined NDI.132–135 More than 200 AVPR2 mutations have been described including missense, nonsense, insertions, deletions, and complex rearrangements.136 Mutations have been categorized into five classes based on mechanism, including abnormal transcription, mRNA processing, translation, aberrant folding and intracellular retention, loss of the G protein binding site, loss of the AVP binding site, and defects in intracellular trafficking.131,137,140 Some patients with X-linked NDI are responsive to high doses of desmopressin. Autosomal recessive NDI (ARNDI) is caused by loss-of-function mutations in the gene for the aquaporin-2 water channel and accounts for about 10% of genetic forms of NDI.137,139 More than 40 known mutations cause ARNDI. Autosomal dominant forms of NDI are also caused by mutations in AQP2 that are functional but fail to be transported to the apical membrane. They account for < 1% of genetic forms of NDI and generally have a milder phenotype than ARNDI or X-linked NDI.
Gain-of-function mutations in the V2 vasopressin receptor have also been reported.141 DNA sequencing of two patients’ V2R gene identified heterozygous missense mutations in both, with resultant changes in codon 137 from arginine to cysteine (R137C) or leucine (R137L). These mutations resulted in constitutive activation of the receptor and clinical features of inappropriate antidiuretic hormone secretion (SIADH), which was termed nephrogenic syndrome of inappropriate antidiuresis (NSIAD).141 The patient with the R137L mutation demonstrated the expected decrease in AVP levels with a water-loading test but urine aquaporin 2 levels remained inappropriately elevated.142
The Glycoprotein Hormone Receptor Group
The glycoprotein hormones include TSH, FSH, LH, and HCG. These hormones share common α subunits that dimerize with hormone-specific β subunits. TSH, FSH, and LH bind to the extracellular N-terminal domain of the TSH, FSH, and LH receptors, respectively.1,3,143,144 The effects of HCG are mediated by the LH receptor, which is also known as the luteinizing hormone/choriogonadotropin receptor (LHCGR).145
Glycoprotein hormone receptors have a large (350 to 400 residues) extracellular N-terminal domain, also known as the ectodomain, that participates in ligand binding (see Figure 3-3).4,145 The ectodomain includes leucine-rich repeats that are highly conserved among the glycoprotein hormone receptors.4,145 There is 39% to 46% similarity of the ectodomain and 68% to 72% similarity of the transmembrane or serpentine domain among the three glycoprotein hormone receptors.4
The glycoprotein hormone receptors are coupled to Gs, and hormone binding stimulates adenylyl cyclase, leading to increased intracellular cAMP levels and protein kinase A (PKA) activation.145 Mutations leading to endocrine dysfunction have been reported for each of the glycoprotein hormone receptors.
LHCGR Receptors
Both inactivating and activating mutations of the LH receptor have been found in humans.145 The LH receptor gene is located in chromosome 2 p21 and consists of 11 exons.146,147 Exon 1 encodes a peptide that directs the LH receptor to the plasma membrane.145 Exons 2 through 10 encode the ectodomain.145 The last exon encodes the transmembrane domains that are also known as the serpentine regions.4,145,146 Single nonsense mutations, amino acid changes, and partial gene deletions have been described that generate LH receptors with decreased activity.145 Single–amino-acid changes have also been found that lead to activation of Gs in the absence of ligand binding.145
Development of LH resistance requires biallelic mutations that inactivate the LH receptor gene, as one normal receptor allele is capable of producing adequate receptor protein to ensure physiologic signaling.145 In contrast, activating mutations of the LH receptor gene cause endocrine disorders in the heterozygous state.145
In the fetus, LH receptors are primarily activated by HCG.145 Leydig cells begin to express LH receptors shortly after testicular differentiation at 8 weeks of gestation.145 Thereafter, androgen production due to activation of these receptors by HCG plays an important role in the development of male genitalia and testicular descent.145 Thus, male infants with inactivating mutations of the LH receptor may present with abnormally developed genitalia—including micropenis, cryptorchidism, and an XY disorder of sexual differentiation.145
Males with mutations that completely inactivate the LH receptor exhibit failure of fetal testicular Leydig cell differentiation. This phenotype, which is known as type 1 Leydig cell hypoplasia, includes female external genitalia with a blind-ending vagina, absence of Müllerian derivatives, and inguinal testes with absent or immature Leydig cells.148–155 In addition, patients have elevated serum LH levels, normal serum FSH levels, and decreased serum testosterone levels that do not increase in response to HCG administration.148–155 Mutations that lead to this phenotype include a nonsense mutation (Arg545Stop) that results in a receptor that is missing TM4-7, an Ala593Pro change, and a TM7 deletion (ΔLeu608, Val609) that decreases cell surface expression of the LH receptor.153,154,156 These mutant receptors are unable to couple to Gs.153,154,156
Males with mutations that do not completely inactivate the LH receptor present with type 2 Leydig cell hypoplasia, which is characterized by a small phallus and decreased virilization.152 A mutation that leads to this phenotype includes the insertion of a charged lysine at position 625 of TM7 in place of hydrophobic isoleucine that disrupts signal transduction.157 Another mutation (Ser616Tyr, found in patients with mild Leydig cell hypoplasia) is associated with decreased cell surface expression of the LH receptor.154,157 Other deletion and nonsense mutations have also been found to cause mild Leydig cell hypoplasia.145
Males with inactivating mutations of the LH receptor may also present with a phenotype intermediate in severity between type 1 and type 2 Leydig cell hypoplasia. A compound heterozygote patient with Ser616Tyr on one allele and an inactivating deletion (Δexon 8) on the other allele presented with Leydig cell hypoplasia, micropenis, and hypospadias.158 The Cys131Arg mutation has also been found in patients with Leydig cell hypoplasia, small phallus, and hypospadias.159 This mutation is located in the leucine-rich repeat segment of the LH receptor extracellular domain and interferes with high-affinity ligand binding.159
Deletion of exon 10 of the LH receptor gene leads to an LH receptor that binds LH and HCG normally.160 Interestingly, whereas HCG binding can elicit normal transmembrane signaling, LH binding fails to activate the receptor.160 Because HCG is the principal in utero LH receptor-activating hormone, and second-messenger response of the mutant receptors to HCG is not impaired, it is not surprising that a male patient found to be homozygous for the mutation was born with normal male genitalia.62,160 Pubertal progression and later gonadal function, however, are dependent on LH activation of the LH receptor.62,160
Because deletion of exon 10 of the LH receptor gene results in a mutant LH receptor with diminished intracellular signaling in response to LH, it is also not surprising that the patient homozygous for this mutation was found to have delayed pubertal development, small testes, and hypergonadotropic hypogonadism when evaluated at the age of 18 years.160 Prolonged HCG therapy resulted in normalization of testicular testosterone production, increased testicular size, and the appearance of spermatozoa in semen.160 Similarly, inactivating mutations of the LH β subunit cause abnormal pubertal development, severe testosterone deficiency and azoospermia but normal external genitalia in males. In females inactivating mutations of the LH β subunit are associated with normal pubertal development and menarche followed by oligomenorrhea, enlarged multicystic ovaries and infertility.161
Mutations that constitutively activate LH receptors cause male-limited precocious puberty (MLPP), also known as testotoxicosis—which may be familial or sporadic.155,162,163 Boys with this condition present with GnRH-independent precocious puberty before the age of 4 years when the Asp578Gly is present and as early as the first year of life when the Asp578Tyr mutation is present.145,164–166 Patients with this condition may also have an enlarged phallus at birth.164
During the first 5 years of life, patients with MLPP have very low LH and FSH levels but have testosterone levels in pubertal range.167 During adolescence and adult life, testosterone levels do not increase above age-appropriate concentrations and gonadotropin levels normalize.145,167–169 Thus, adolescents and adults with MLPP do not usually manifest signs of androgen excess (such as hirsutism or severe acne).145,167 Most mutations that cause MLPP are located in the TM6 and i3, regions that participate in receptor-Gs protein coupling.145 A milder phenotype was reported in a patient with a heterozygous activating mutation (C617Y) in TM7.170 This mutation was inherited from the patient’s mother who was apparently unaffected. Somatic activating mutations cause sporadic Leydig cell adenomas.105,171
Activating mutations of the LH receptor do not appear to have a phenotype in females. In prepubertal girls, this may be due to low or absent LH receptor expression or due to insufficient aromatase expression in prepubertal granulosa cells. During puberty, activation of LH receptors on ovarian theca cells leads to the production of androgens that are converted to estrogens by aromatase in granulosa cells.145 LH, along with FSH, also plays a role in inducing the differentiation of follicles into Graafian follicles and triggers ovulation and release of the oocyte.145 Detailed phenotyping of the carrier mother of a MLPP male with the Asp578Gly activating mutation of the LH receptor failed to reveal any abnormalities in her menstrual cycles or fertility. LH dynamics, androgen, and FSH levels as well as response to GnRH agonists were normal.173
Females with inactivating mutations of the LH receptor may be asymptomatic or present with primary amenorrhea.145 Females with complete inactivating LH receptor mutations may present with primary amenorrhea, inability to ovulate, and decreased estrogen and progesterone levels accompanied by elevated LH and FSH levels.154,174 Affected individuals may have signs of low estrogen levels, including a hypoplastic uterus, a thin-walled vagina, decreased vaginal secretions, and decreased bone mass.154,174 A homozygous LH receptor mutation (N400S) has been associated with empty follicle syndrome.
FSH Receptors
Inactivating and activating FSH receptor mutations have also been described,175 but they are far less common than LH receptor mutations.175 The FSH receptor gene is located in chromosome 2 at p21 and contains 10 exons.176 The last exon of the FSH receptor gene encodes the transmembrane and intracellular domains.177
FSH is required in females for normal follicle maturation and the regulation of estrogen production by ovarian granulosa cells.175,178,179 FSH is required in pubertal males for Sertoli cell proliferation, testicular growth, and the maintenance of spermatogenesis.175,180
The first inactivating mutation of the FSH receptor was found in Finnish females with autosomal recessive inherited hypergonadotropic ovarian dysgenesis (ODG). ODG is characterized by primary amenorrhea, infertility, and streak or hypoplastic ovaries in the presence of a 46XX karyotype and elevated gonadotropin levels.181 Twenty-two out of 75 Finnish patients with ODG were found to be homozygous for a C566T point mutation in exon 7 of the FSH receptor gene.182 This mutation leads to the production of an FSH receptor with an Ala189Val substitution in an area of the extracellular ligand- binding domain that is thought to play a role in turnover of the receptor or in directing the receptor to the plasma membrane.182 The mutated receptor demonstrates normal ligand-binding affinity but has decreased binding capacity and impaired signal transduction when studied in transfected mouse Sertoli cells.182 Males homozygous for this mutation have variable impairment of spermatogenesis and low to low-normal testicular volume but are not azoospermic and can be fertile.183 The C556T point mutation is uncommon outside Finland, where the carrier frequency is 0.96%.184 Other mutations that alter signal transduction but not receptor expression or binding include Ala189Val, Asn191Ile, Ala419Thr, and Phe591Ser. The Ala189Val mutation causes primary hypergonadotropic amenorrhea in women and no spermatogenesis in men in the homozygous state and secondary amenorrhea in the heterozygous state.185,186 The nearby Asn191Ile mutation also causes hypergonadotropic amenorrhea in the homozygous state but no clinical phenotype in the heterozygous state.187 The Ala419Thr mutation was identified in a heterozygous woman with primary amenorrhea.188 The Phe591Ser mutation causes primary amenorrhea and premature ovarian failure (POF) in the homozygous state and a predisposition to sex cord ovarian tumors in the heterozygous state.189 Primary amenorrhea and POF have been described in women with homozygous mutations that totally impaired receptor binding to FSH190 or that resulted in reduced expression of the FSH receptor on the cell surface.191
Compound heterozygosity for mutations that cause partial loss of FSH receptor function may cause endocrine dysfunction in women.192,193 Women may present with infertility, secondary amenorrhea, osteoporosis, and a history of delayed onset of puberty accompanied by elevated LH and FSH, low-normal plasma estradiol, low plasma inhibin B levels, slightly enlarged ovaries with immature follicles, and a small uterus.192 This may be caused by FSH receptor gene mutations that result in an Ile160Thr mutation in the extracellular domain that impairs cell surface expression and an Arg573Cys mutation in e3 that interferes with signal transduction.192 Other women present with primary amenorrhea and very elevated gonadotropin, low plasma estradiol and inhibin B levels, normal-size ovaries with immature follicles, and a normal-size uterus.193 This condition is associated with an Asp224Val substitution in the extracellular domain leading to impaired cell-surface expression and a Leu601Val substitution in e3 impairing signal transduction.193
Activating mutations of the FSH receptor have also been described. Surprisingly, a hypophysectomized male was found to be fertile and to have serum testosterone levels above 4.9 nmol/L and normal testis volume in spite of undetectable gonadotropin levels.194 This patient was found to be heterozygote for an A1700G mutation in exon 10 of the FSH receptor gene that resulted in an Asp567Gly substitution in an area of the third intracytoplasmatic loop that is highly conserved among FSH, LH, and TSH receptors.194–196 The same substitution in corresponding areas of the LH and TSH receptors also results in constitutively active receptors and is found in MLPP and thyroid adenomas, respectively.194–196 Other activating mutations have been identified to cause spontaneous ovarian hyperstimulation syndrome (OHSS). Ovarian hyperstimulation syndrome is a common complication of treatment protocols used to induce ova for in vitro fertilization and is characterized by multiple follicular cysts lined by luteinized cells, which can result in abdominal discomfort and distention as well as ovarian enlargement and fluid sequestration. One such mutation is the Asp567Asn, which was found in a woman with recurrent spontaneous OHSS.197 The Thr449Ile and Thr449Ala mutations cause a conformational change that leads to loss of specificity for FSH leading to sensitivity to HCG198 and TSH199 causing spontaneous OHSS during pregnancy or with hypothyroidism. The Ile545Thr mutation caused spontaneous OHSS in a woman during the first trimester of pregnancy despite a normal HCG level.200 This mutant receptor displayed detectable constitutive activity as well as promiscuous activation by HCG and TSH.
TSH Receptors
The TSH receptor gene is located on chromosome 14 and contains 10 exons, with the first nine exons encoding the large extracellular domain and the tenth exon coding the remainder of the receptor.201–204 At low extracellular TSH concentrations, TSH receptor activation leads to stimulation of Gαs—which activates adenylyl cyclase, resulting in increased intracellular cAMP levels.205,206 At higher extracellular TSH concentrations, activation of the TSH receptor also stimulates the Gq and G11 proteins—activating phospholipase C and resulting in the production of diacylglycerol and inositol phosphate.206
TSH receptors differ from the other glycoprotein hormone receptors in that they exist in two equally active forms.207,208 These are the single-chain and two-subunit forms of the TSH receptor (Figure 3-4). The single-chain form of the TSH receptor is made up of three contiguous subunits: the A subunit, C peptide, and B subunit.208–210 The A subunit begins at the N terminal of the extracellular domain and contains most of the extracellular domain.208–210 The C peptide is connected to the C terminal of the A subunit and continues the extracellular domain.208–210 The C peptide contains a 50-amino-acid sequence that is only found in TSH receptors.208–210 The B subunit is connected to the C terminal of the C peptide and contains the TMs and the C-terminal cytoplasmic portion of the receptor.208–210 The two-subunit form of the receptor is missing the C peptide, which is cleaved from the protein during intracellular processing and consists of the A and B subunits attached by disulfide bonds.211–214 It is surprising that both receptor forms are activated equally by TSH because the C peptide and nearby regions of the A and B subunits participate in signal transduction.207–209,215,216
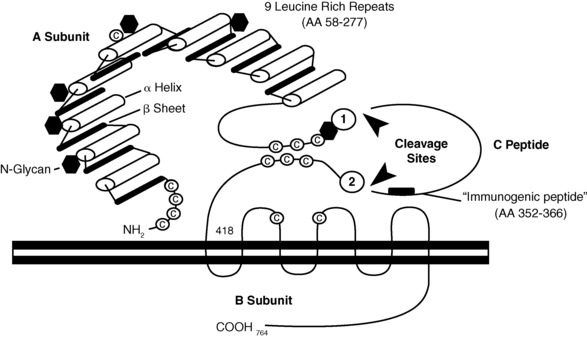
FIGURE 3-4  The TSH receptor. There are two forms of TSH receptors. The single-chain form is made up of an A subunit, C peptide, and B subunit. Posttranslational cleavage of the C peptide from the single chain form results in the two-subunit form. This form consists of the A subunit joined to the B subunit by disulfide bonds between the C-terminal cysteine residues of the A subunit and the N-terminal cysteine residues of the B subunit. (Reproduced with permission from Rapoport, B., Chazenbalk, G. D., Jaume, J. C., & McLachlan, S. M. (1998). The thyrotropin [TSH] receptor: interaction with TSH and autoantibodies. Endocr Rev, 19, 676. Copyright 1998, The Endocrine Society.)
The TSH receptor. There are two forms of TSH receptors. The single-chain form is made up of an A subunit, C peptide, and B subunit. Posttranslational cleavage of the C peptide from the single chain form results in the two-subunit form. This form consists of the A subunit joined to the B subunit by disulfide bonds between the C-terminal cysteine residues of the A subunit and the N-terminal cysteine residues of the B subunit. (Reproduced with permission from Rapoport, B., Chazenbalk, G. D., Jaume, J. C., & McLachlan, S. M. (1998). The thyrotropin [TSH] receptor: interaction with TSH and autoantibodies. Endocr Rev, 19, 676. Copyright 1998, The Endocrine Society.)
Spontaneous single-allele mutations of the TSH receptor gene leading to replacement of Ser-281 (near the C terminal of the A subunit, with Ile, Thr, or Asn) result in a constitutively active TSH receptor that may cause intrauterine or congenital hyperthyroidism, or toxic adenomas.210,217–219 Activating somatic mutations that cause toxic adenomas have also been found in different transmembrane domains of the TSH receptor.220–227 More specifically, clusters of mutations are located in the i3 and TM6 regions—found to be involved with signal transduction in all glycoprotein hormone receptors.220–222,224–226 The prevalence of activating mutations of the TSH receptor in toxic adenomas has been estimated to range from 2.5% in Japan to 86% in Brazil.223,224,226,228–231
Activating somatic mutations of the TSH receptor have also been found in multinodular goiters.232 Interestingly, different activating mutations have been found in separate nodules in the same individual.232 Some well-differentiated thyroid carcinomas have activating mutations of the TSH receptor.233–235


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


