David C. Hooper, Jacob Strahilevitz
Quinolones
The first member of the quinolone class of antimicrobial agents, nalidixic acid, is a 1,8-naphthyridine structure that was identified by Lesher and associates in 1962 among the byproducts of chloroquine synthesis. Oxolinic acid and cinoxacin were also developed in the 1970s, but it was the identification in the 1980s of the fluorine- and piperazinyl-substituted derivatives with substantially greater potency and expanded spectrum that began a resurgence in development and a rapid and steady expansion of this class of compounds. A broad spectrum of activity, good oral absorption, and generally good overall tolerability have resulted in extensive clinical use of the newer fluoroquinolones. Several quinolones, however, including temafloxacin, sparfloxacin, grepafloxacin, trovafloxacin, and gatifloxacin, were removed from clinical use after approval because of toxicities, which were uncommon but severe in some cases. This chapter focuses on those quinolones that are currently in clinical use.
Chemical Structures
All current quinolone derivatives in clinical use have a dual ring structure with a nitrogen at position 1, a carbonyl group at position 4, and a carboxyl group attached to the carbon at the 3 position of the first ring (Fig. 34-1). Several different dual ring structures (cinnoline [nitrogens at positions 1 and 2], pyridopyrimidine [nitrogens at positions 1, 6, and 8], and 2-pyridone [a dual ring structure with the nitrogen located at the junction of the two rings]) have been developed, but quinolones, which themselves have a carbon at position 8 in the second ring, and naphthyridines, which contain a nitrogen at position 8, have been most widely successful. Both quinolones and naphthyridines, however, are commonly referred to as quinolones.
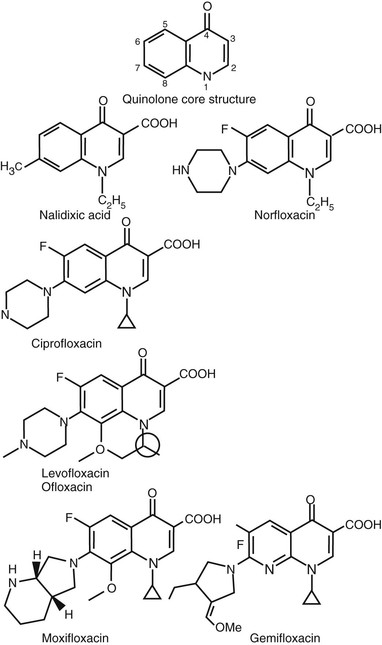
Nalidixic acid is a 1,8-naphthyridine with 1-ethyl and 7-methyl substituents (see Fig. 34-1). Oxolinic acid (quinolone ring; see Fig. 34-1) and cinoxacin (cinnoline ring; not shown in Fig. 34-1) also have 1-ethyl substitutions, as well as a dioxolo ring bridging positions 6 and 7. Potency is greatly improved by the addition of a fluorine at position 6, and potency against gram-negative bacteria is further enhanced by the addition of a piperazinyl (norfloxacin, enoxacin, ciprofloxacin), methyl-piperazinyl (pefloxacin, ofloxacin, lomefloxacin, fleroxacin, temafloxacin, levofloxacin, grepafloxacin, gatifloxacin), or dimethyl-piperazinyl (sparfloxacin) substituent at position 7.1 Methyl substituents on the piperazine ring generally result in improved oral bioavailability. These structural features are common to most of the newer quinolone derivatives now in clinical use. Pyrrolidinyl (tosufloxacin, clinafloxacin, gemifloxacin) or dual ring substituents (trovafloxacin, moxifloxacin, sitafloxacin) at position 7 enhance activity against gram-positive bacteria. A number of compounds (sparfloxacin, gatifloxacin, moxifloxacin, gemifloxacin) use the 1-cyclopropyl group, which enhances potency, particularly against gram-negative bacteria, and was originally identified for ciprofloxacin. The 1-difluorophenyl group found in temafloxacin, tosufloxacin, and trovafloxacin adds potency against gram-positive bacteria. An additional ring structure bridging positions 1 and 8 is found in ofloxacin and levofloxacin; the additional ring of ofloxacin contains an asymmetrical carbon, resulting in stereoisomeric forms. Thus, ofloxacin is a racemic mixture of the two stereoisomers, one of which is responsible for most of the potency against bacteria and purified DNA gyrase. Levofloxacin is the more potent of the ofloxacin stereoisomers and thus predictably has twice the potency of ofloxacin in vitro. At position 5, replacement of the hydrogen by an amino group (sparfloxacin) or a methyl group (grepafloxacin) results in some enhancement of activity against gram-positive bacteria. At position 8, addition of a halide (chlorine-clinafloxacin, fluorine-sparfloxacin, and sitafloxacin) or a methoxy group (gatifloxacin, moxifloxacin) enhances activity against anaerobic bacteria. Halides at position 8 increase the risk of phototoxicity, but methoxy groups at this position reduce risks of phototoxicity, even relative to compounds with a hydrogen at position 8.
Coming full circle in structural modifications, most recently, newer “desfluoro” quinolones (garenoxacin) have been identified with excellent and similar potency to their fluorinated counterparts but with possible reductions in joint toxicities.2–4
Mechanism of Action
The quinolones rapidly inhibit bacterial DNA synthesis, an event that is followed by rapid bacterial cell death. The molecular events that underlie these actions are understood in part, but details remain to be defined.5
Quinolones inhibit the enzymatic activities of two members of the topoisomerase class of enzymes—DNA gyrase and topoisomerase IV—and promote the cleavage of DNA in these enzyme-DNA complexes. DNA gyrase, which was the first-recognized target of quinolones, is an essential bacterial enzyme composed of two A and two B subunits, products of the gyrA and gyrB genes, respectively.6,7 DNA gyrase uniquely catalyzes the introduction of negative superhelical twists into closed covalently circular chromosomal and plasmid DNA within the bacterial cell. The superhelical state of intracellular DNA is regulated by the actions of DNA gyrase and topoisomerase I, which removes DNA superhelical twists but is not inhibited by quinolones. DNA superhelicity affects the initiation of DNA replication and transcription of many genes. DNA gyrase is also responsible for removing positive superhelical twists that accumulate ahead of the DNA replication fork. These activities result from the enzyme’s coordinated breaking of both strands of duplex DNA, passage of another segment of DNA through the break, and resealing of the break, a mechanism that defines type II topoisomerases.
Quinolones also inhibit the activities of topoisomerase IV, another type II topoisomerase that is composed of two subunits encoded by the parC and parE genes. Topoisomerase IV and DNA gyrase are structurally related; parC is homologous to gyrA, and parE is homologous to gyrB.8 Topoisomerase IV functions to resolve (decatenate) interlinked (catenated) daughter DNA molecules that result from replication of circular DNA, to allow their segregation into daughter cells. Thus, DNA gyrase and topoisomerase IV have distinct essential roles in bacterial DNA replication.9 Although gyrase can mediate the functions of topoisomerase IV (albeit less efficiently as a decatenase), topoisomerase IV lacks the ability to introduce negative supercoils into DNA, a function that is unique to DNA gyrase. A few species of human pathogens (e.g., Mycobacterium tuberculosis, Treponema pallidum) lack topoisomerase IV, and in the case of M. tuberculosis, gyrase appears to serve the decatenation function of topoisomerase IV, in addition to its own functions.10
Quinolones appear to function by trapping or stabilizing the enzyme-DNA complexes after strand breakage and before resealing of DNA. This trapped complex appears to function as a cellular poison, possibly by generating a DNA break that the cell is able to repair only poorly.5,11 Quinolones have been shown to bind specifically to the complex of DNA gyrase and DNA rather than to DNA gyrase alone.12,13 The importance of the interaction of quinolones with the gyrase-DNA complex for the antibacterial activities of the quinolones in Escherichia coli is indicated by the identification of bacterial single gyrA or gyrB mutants that are resistant to quinolone enzymatic inhibition and produce gyrase-DNA complexes with reduced quinolone binding (see “Mechanisms of Acquired Bacterial Resistance”).13 In contrast to the information for E. coli, which is also similar to that for other gram-negative bacteria studied, for Staphylococcus aureus and Streptococcus pneumoniae, interactions of quinolones with topoisomerase IV have been shown to determine antibacterial activity by the identification of parC (initially named grlA in S. aureus) and parE (grlB in S. aureus) single mutants that have reduced quinolone activity.14 Based on studies of mutants of this type, a general pattern has emerged. For most gram-negative bacteria, DNA gyrase is the primary quinolone target, and for many gram-positive bacteria, topoisomerase IV is the primary target, with gyrase being the secondary target. There are exceptions, however, that depend on the quinolone studied.15,16 These patterns appear to result from the relative sensitivities of these two topoisomerases to a given quinolone, with the more sensitive of the two enzymes defining the primary target of a particular quinolone.17 Some quinolones under development appear to have similar potencies against both DNA gyrase and topoisomerase IV in some bacterial species.18,19
Quinolone inhibition of bacterial DNA replication and bacterial killing may be dissociated under some conditions, suggesting that events in addition to the initial interaction of quinolones with the topoisomerase-DNA complex may be required for cell killing.20 In particular, inhibitors of RNA and protein synthesis reduce the bactericidal activity of some quinolones but do not affect their ability to inhibit bacterial DNA synthesis.21 Thus, inhibition of bacterial DNA synthesis per se is not sufficient to account for bacterial killing, and possibly, newly synthesized gene products may also be necessary. This effect may account for the observations that at high concentrations of quinolones, which also secondarily inhibit protein synthesis, cell killing is reduced.5,20
The nature of the gene products (in addition to DNA gyrase and topoisomerase IV) that contribute to killing, however, has yet to be defined. The gene products in the RecA-SOS DNA repair and recombination system, the expression of which is known to be induced by the damage to bacterial DNA caused by quinolones and other agents, appear to function at least in part to repair quinolone-induced DNA damage because rec mutants with defective function are hypersusceptible to killing by quinolones.22 Certain hip mutants exhibit a higher fraction of persisters or surviving cells after treatment with bactericidal antimicrobial agents, without exhibiting major differences in the concentrations of drug that inhibit bacterial growth. hipA mutants selected for reduced killing by ampicillin have alterations in the cell wall and are also killed less well by quinolones, and hipQ mutants selected for reduced killing by quinolones also exhibit selective reduced killing by β-lactams.23,24 The function of the hipA and hipQ gene products is not known, but the overlap in reduced killing by both β-lactams and quinolones suggests that after the interaction of these classes of drugs with their different targets, there may be common overlapping pathways that are necessary for bacterial lethality. Quinolones, β-lactams, and aminoglycosides have all been shown to generate the production of hydroxyl radicals through a proposed complex series of events involving stimulation of oxidation of reduced nicotinamide adenine dinucleotide via electron transport, which generates superoxide radicals that then damage intracellular thiol-bound iron clusters, releasing ferrous iron for oxidation by the Fenton reaction). Production of hydroxyl radicals can damage cellular macromolecules and contribute to bacterial cell death, thus suggesting one component of a common pathway of bacterial lethality for different classes of bactericidal antibiotics.25
Differences among quinolones in the magnitude of bacterial killing in the presence of rifampin (an inhibitor of RNA synthesis), chloramphenicol (an inhibitor of protein synthesis), phosphate-buffered saline, or anaerobiasis have led to the suggestions that some quinolones may have more than one mechanism of killing.20,26 The molecular events underlying these phenomena, however, remain poorly understood, except that bacterial killing is dependent on drug interaction with complexes of DNA gyrase and topoisomerase IV with DNA5 and is associated with the release of constraints on the DNA breaks in the bacterial chromosome generated by these drug-enzyme-DNA complexes.20
Eukaryotic cells also contain topoisomerases, and eukaryotic topoisomerase II, which is a homodimeric enzyme that is a member of the type IIA class of topoisomerases, such as DNA gyrase and topoisomerase IV, has a similar domain structure and limited primary amino-acid sequence homology to the bacterial enzymes.27–29 Current antibacterial quinolones in clinical use have only minimal activity against mammalian topoisomerase II,30 but other quinolone structures, containing a 7-hydroxyphenyl substituent or an isothiazolo ring bridging positions 2 and 3, have been shown to have substantially enhanced potency against the mammalian enzyme.28
Mechanisms of Acquired Bacterial Resistance
Bacteria acquire resistance to quinolones by spontaneously occurring mutations in chromosomal genes that either alter the target enzymes, DNA gyrase and topoisomerase IV, or alter drug permeation across the bacterial cell membranes.31 Recently, several plasmid-mediated quinolone resistance mechanisms have been identified in clinical isolates of Enterobacteriaceae. These horizontally acquired genes are not sufficient to confer clinical resistance to fluoroquinolones but enable survival under drug exposure and facilitate selection of chromosomal mutations.32 The products of the plasmid-encoded qnr genes have been shown to protect DNA gyrase and topoisomerase IV from quinolone action.33,34 A modification of a common plasmid-encoded aminoglycoside acetylating enzyme, Aac(6′)-Ib-cr, mediates quinolone resistance through acetylation of the nitrogen on the piperazinyl substituent at position 7 of ciprofloxacin and norfloxacin.35 Genes encoding efflux pumps, such as QepA36,37 and OqxAB,38 that include quinolones in their substrate profiles have also been found on plasmids, albeit infrequently to date.
Resistant chromosomal mutants may be selected in the laboratory by plating bacteria on drug-containing agar. The frequency of occurrence of spontaneous mutants differs with the selecting drug concentration and the drug. For gram-negative bacteria selected with the newer fluoroquinolones, frequencies range, in general, from 10-6 or higher at twofold above the minimal inhibitory concentration (MIC) to undetectable (<10-10) at 16- to 32-fold above the MIC. With a similar selection with nalidixic acid, mutants are detected more frequently when selected at a similar factor greater than the MIC because single mutations can cause a higher level of increase in resistance (>30-fold) relative to ciprofloxacin (eightfold) and other fluoroquinolones. This difference results in part because the magnitude of the increase in resistance conferred by a single-target mutation is modified by the interaction of drug with the second target enzyme. For example, for nalidixic acid, which has little activity against topoisomerase IV, a common mutation in the GyrA subunit of DNA gyrase causes a 30-fold increase in MIC. In contrast, for ciprofloxacin, which has activity against topoisomerase IV (albeit less than its activity against gyrase), the same gyrA gene mutation causes only an eightfold increase in MIC, despite the fact that both drugs exhibit similar loss of activity against purified DNA gyrase reconstituted with the resistant GyrA subunit.39 Thus, quinolone interaction with a second target enzyme puts a ceiling on the magnitude of the increase in resistance caused by mutation in the first enzyme target. For some quinolones that have similar potency against both target enzymes, frequencies of selection of first-step mutants may be particularly low because single-target mutations produce little or no increment in resistance.40 Thus, for such quinolones, mutations in both targets are needed to produce substantial increments in MIC. Serial passage of bacteria with increasing concentrations of quinolones selects mutants with high levels of resistance resulting from the additive effects of several mutations involving both enzyme targets.41
Alterations in the A subunit of DNA gyrase that cause quinolone resistance have now been defined in a substantial number of clinical and laboratory isolates of E. coli. These alterations are clustered between amino acids 67 and 106 in the amino terminus of the A protein near the active site of the enzyme (tyrosine-122).31 In particular, changes in serine-83 (to leucine or tryptophan) are most common and cause the largest increment in resistance, as well as reduced binding of drug to the gyrase-DNA complex in vitro. Leucine-83 causes a 128-fold increase in resistance to nalidixic acid but lesser increases in resistance to the newer fluoroquinolones (16- to 32-fold), thus likely accounting for the greater ease of selection of resistant mutants with nalidixic acid. Similar changes in the A subunit have been associated with resistance in many species of gram-negative bacteria. Single amino-acid changes in the midportion of the gyrase B protein have also been found to cause lower levels of resistance to nalidixic acid and fluoroquinolones.31,42
Resistance mutations in the parC gene of topoisomerase IV in S. aureus and S. pneumoniae have been most commonly found at position 80, in which a wild-type serine (homologous to serine-83 of DNA gyrase) is replaced by phenylalanine or tyrosine.14,43 These mutations cause eightfold increases in resistance to several fluoroquinolones. Resistance mutations have also been found less commonly in the parE gene, often in positions similar to those of resistance mutations gyrB.31
The quinolone resistance-determining region is probably larger than previously thought. New quinolones can select in vitro for novel mutations that are responsible, in part, for the resistance phenotype.44 Of interest, sampling soil flora thought to lack prior exposure to fluoroquinolones identified unfamiliar mutational changes in the gyrA gene. However, their role in resistance has not yet been vigorously proven.45
Stepwise increasing resistance occurs by sequential mutations in the gyrA (or gyrB) and parC (or parE) genes, with the first target mutation occurring in a gene for the more sensitive target enzyme. The level of susceptibility of the first-step mutant is then usually determined by the intrinsic level of sensitivity of the secondary target enzyme. A second mutation in a gene for the secondary target then further increases resistance to the level that is determined by the already mutant primary enzyme target. This finding implies that the drug concentration above which two mutations will be required to select resistance will decrease to the extent that the similarity of sensitivities of DNA gyrase and topoisomerase IV to given quinolone approach the same value. In the most highly resistant clinical strains of both gram-positive and gram-negative bacteria, one or more mutations in each of gyrA and parC have been found commonly. Some species, including M. tuberculosis, Helicobacter pylori, and T. pallidum, appear to lack genes for topoisomerase IV.31 Thus, gyrase or another enzyme may carry out the functions of topoisomerase IV in these organisms, and target resistance may occur more readily in the absence of a second drug target to limit the effects of resistance mutations in gyrase.
The routes of quinolone permeation across bacterial cell membranes are not fully defined, but the hydrophilic congeners appear to diffuse across the gram-negative bacterial outer membrane through porin channels. In E. coli and Pseudomonas aeruginosa, resistance mutations in genes that affect expression of outer membrane proteins have been described.46,47 In both cases, resistance cannot be explained by reduced diffusion alone, and reduced drug accumulation in some mutants is energy dependent, being abolished by agents that collapse the proton gradient across the membrane. In E. coli, resistance of multiple antibiotic resistance (Mar) mutants, which exhibit reduced porin channels, is dependent on the AcrAB efflux pump, which is linked to the TolC outer membrane protein.48,49 Overexpression of AcrAB has also been associated with quinolone resistance in clinical isolates.50 In P. aeruginosa, resistance has been shown to be caused by increased expression of one of several sets of three genes that appear to encode an efflux pump in the inner membrane, a periplasm-spanning membrane fusion protein, and a linked outer membrane protein (e.g., MexAB-OprM, composed of the MexA membrane fusion protein, MexB inner membrane efflux pumps, and OprM outer membrane protein),31,46,51 and such increased expression of one or more pump complexes is found commonly along with gyrA mutations in quinolone-resistant clinical isolates of P. aeruginosa.52 Regulation of expression appears to be complex, and overexpression has in some cases been shown to be due to mutations in specific regulators. Resistance in many mutants of this type is pleiotropic, with additional low levels of resistance to tetracycline, chloramphenicol, and some β-lactams, because of the broad substrates profiles of most such pumps; hence, the term multidrug resistance (MDR) is often applied to these efflux pumps.46,53 For an extensive review of clinically relevant chromosomally encoded MDR efflux pumps see the review by Piddock.53
In gram-positive bacteria, which lack an outer membrane, overexpression of endogenous efflux pumps has also been shown to cause low-level quinolone resistance. The S. aureus norA gene encodes a native membrane protein that pumps hydrophilic quinolones, driven by the proton gradient across the cell membrane,54 and overexpression of NorA, because of a mutation in the norA promoter region or in other regulators, causes resistance to norfloxacin, ciprofloxacin, and levofloxacin, in order of decreasing magnitude of the effect on MICs.55 Some quinolones, such as moxifloxacin, are not affected by NorA overexpression. Among the more recently discovered pumps, NorB and NorC can cause resistance to these quinolones as well.56,57 In S. pneumoniae, PmrA, a pump with a structure similar to NorA, can also contribute to reduced quinolone susceptibility.58 Reserpine, an inhibitor of several efflux pumps in gram-positive bacteria, improves MICs of some quinolones in clinical isolates of S. pneumoniae and viridans streptococci.59 More recently, a PatA/PatB heterodimer was shown to form an ABC transporter that confers resistance to norfloxacin and ciprofloxacin in S. pneumoniae.60 It is likely that further study of potential MDR pumps in S. aureus, S. pneumoniae, and other gram-positive bacteria, aided by the increasing number of species with completely sequenced genomes, will identify additional quinolone resistance mechanisms resulting from overexpression of other as yet undefined MDR efflux pumps.
MDR efflux pumps of this type appear to be present in most, if not all, bacteria, and in many cases multiple pumps, which may vary in their relative levels of expression under different conditions of growth or antibiotic exposure and selection, are present in a given species. For example, in P. aeruginosa, the MexAB-OprM pump complex is normally expressed, but selection with different quinolones can result in mutants that overexpress the MexCD-OprJ or MexEF-OprN pump complexes, presumably reflecting the particular ability of the overexpressed pump to remove the selecting quinolone from the cell.61 A similarly complex picture of expression of various efflux pumps is also emerging in gram-positive bacteria, and it is likely that regulation of transporter expression is controlled at a number of levels. For example, the global regulator MgrA acts as a positive regulator of NorA62 but as a negative regulator of NorB and NorC.56,57 Moreover, expression of efflux pumps may not necessarily be to extrude the antibiotics. Using the community-acquired methicillin-resistant S. aureus MW2 strain in a murine abscess model, NorB was selectively upregulated in the abscess relative to the expression in laboratory media, and this was associated with improved growth fitness of strain MW2 in the abscess (in the absence of antibiotic exposure).63 Thus, physiologic overexpression of NorB or other drug-resistance pumps in an abscess environment might contribute in part to the reduced responses to antibacterial agents seen during treatment of abscesses, as well as discordance between determinations of bacterial susceptibility in vitro and responses to antimicrobials in vivo. Two plasmid-mediated quinolone efflux pumps have also been found: OqxAB, which confers resistance to the antibiotic olaquindox (a quinoxaline derivative that is used in agriculture as a veterinary growth promoter) as well as an increased MIC to nalidixic acid and ciprofloxacin, and QepA, which mediates increased resistance to several antibiotics, including ciprofloxacin and erythromycin.64,65
Physiologic overexpression of pumps (without regulatory mutations) occurring during infection but not during laboratory growth conditions could also affect outcomes of therapy in a way not necessarily predicted by in vitro methods for susceptibility testing. Compounds that can inhibit all of the known MDR pumps of P. aeruginosa have been identified with the idea of combining such compounds with quinolones or other antimicrobial agents to improve activity in wild-type bacteria and to reduce resistance caused by pump overexpression, a concept analogous to the combination of β-lactams with β-lactamase inhibitors.66 As yet, no such MDR pump inhibitor–containing combinations are available for clinical use.
Plasmid-mediated quinolone resistance, long thought not to occur, was first identified and verified in multidrug-resistant, clinical isolates of K. pneumoniae from Alabama.67 Used as donors, these isolates transferred plasmids by conjugation to a recipient laboratory E. coli with selection for resistance to β-lactams. Unexpectedly, the recipients also acquired low-level resistance to quinolones as well. The plasmid-encoded gene responsible for quinolone resistance, qnr, was located on class I integrons flanked by other resistance genes, which can tranfer multidrug resistance en bloc with the plasmid.33 The gene qnr encodes a protein of the pentapeptide repeat family that is able to protect purified DNA gyrase and topoisomerase IV from quinolone action. How this protection occurs at the molecular level has not yet been defined. The plasmid-borne qnr genes currently comprise five families—qnrA, qnrB, qnrS, qnrC, and qnrD—and include more than 80 alleles.68,69 qnr genes have already been detected worldwide and are found in 1% to 7% of tested Enterobacteriaceae, predominantly in strains of Enterobacter spp., Klebsiella pneumoniae, and E. coli. Although resistance mediated by qnr genes alone is usually low level, qnr plasmids are usually found in strains of Enterobacteriaceae with additional chromosomal resistance mutations, and the presence of qnr has been shown to increase the frequency of selection of these mutations, presumably by reducing the quinolone therapeutic index.
Resistance mediated by a fluoroquinolone-modifying enzyme has also been recently reported. A variant of the gene encoding aminoglycoside acetyltransferase Aac(6′)-Ib was able to reduce the activity of ciprofloxacin by N-acetylation at the amino nitrogen on its piperazinyl substituent.35 The increase in MIC conferred by Aac(6′)-Ib-cr was smaller than that conferred by Qnr proteins, and it was selective only for ciprofloxacin and norfloxacin, as predicted by their chemical structure. Other quinolones lacking an unsubstituted piperazinyl nitrogen were unaffected (see Fig. 34-1). Although the increase in MIC of ciprofloxacin and norfloxacin was modest (threefold to fourfold), the effect on mutant prevention concentration was marked and facilitated selection of resistant clones of wild-type E. coli at 1.6 µg/mL, a level approximating the peak serum concentration of free ciprofloxacin during therapy.35
Infections by salmonellae with gyrA mutations have been associated with poor clinical response among patients receiving ciprofloxacin. Because of the reduced activity of nalidixic acid relative to newer fluoroquinolones against topoisomerase IV, this phenotype can be easily detected by decreased fluoroquinolone susceptibility and nalidixic acid resistance.70,71 The clinical significance of plasmid-mediated quinolone resistance has not been well characterized, and plasmid-mediated quinolone resistance is difficult to detect in the clinical laboratory. The MIC elevation of both nalidixic acid and ciprofloxacin for Qnr-positive strains is modest, and Aac(6′)-Ib-cr does not affect nalidixic acid. Nevertheless, guidelines to clinical laboratories were recently changed to better detect salmonellae that do not carry these resistance mechanisms.72
Antimicrobial Activity
Current quinolones are most active against aerobic gram-negative bacilli, particularly members of the family Enterobacteriaceae and Haemophilus spp., and against gram-negative cocci, such as Neisseria spp. and Moraxella (Branhamella) catarrhalis (Table 34-1).73–76 Relative to nalidixic acid, the fluoroquinolones also have additional activity against gram-negative bacilli, such as P. aeruginosa (see Table 34-1), and against staphylococci (Table 34-2). Ciprofloxacin remains the most potent marketed fluoroquinolone against gram-negative bacteria, and it and levofloxacin in the United States and sitafloxacin in Japan are the only available quinolones with sufficient potency for use against susceptible strains of P. aeruginosa. Resistance may emerge easily, however, when these quinolones are used alone for treatment of serious pseudomonal infections. For norfloxacin, ciprofloxacin, and ofloxacin, activity against streptococci and many anaerobes is limited (see Table 34-2; Table 34-3). Agents released subsequently in the United States—levofloxacin, gatifloxacin (marketed as an ophthalmic solution), moxifloxacin, and gemifloxacin—however, have greater potency against these organisms, with gemifloxacin being especially potent against S. pneumoniae. For the fluoroquinolones that are used for treatment of infections outside the urinary tract, the MICs listed in Table 34-1 should be interpreted in relation to peak drug concentrations in serum, which range from 1.1 to 6.4 µg/mL (with usual dosing), and in relation to drug concentrations in urine that are manyfold higher for most quinolones, except those that are largely excreted by nonrenal mechanisms (see “Pharmacology”). For highly susceptible organisms, MICs may be 10- to 30-fold below achievable serum concentrations. Oral ciprofloxacin and levofloxacin are recommended for prophylaxis of anthrax, with potential similar efficacy and toxicity to doxycycline. For treatment of severe anthrax disease, parenteral fluoroquinolones are favored over doxycycline.77 It should be noted that quinolone resistance can be selected for in Bacillus anthracis. 78 Thus, efficacy may not be predicted in the unfortunate case of use of B. anthracis as an agent of bioterrorism. Fluoroquinolones may have activity against S. aureus isolates, but they are not routinely recommended because resistance may emerge with monotherapy.
TABLE 34-1
Activity of Selected Quinolones against Selected Gram-Negative, Mycoplasmal, and Chlamydial Pathogens in Vitro
| ORGANISM | REPRESENTATIVE MIC90 (range) (µg/mL)*,† | ||||||||
| Nalidixic Acid | Norfloxacin | Pefloxacin | Ciprofloxacin | Ofloxacin | Levofloxacin | Gatifloxacin | Moxifloxacin | Gemifloxacin | |
| Acinetobacter spp. | (32-256) | (8-64) | (1-8) | 0.25-2 (0.25->128) | 0.25->8 | 0.05-32 | 0.03-8 | >0.25-16 | 4-32 (0.008-32) |
| Aeromonas spp. | 0.5 | 0.03 | 0.03 | 0.008-≤0.06 | 0.03-≤0.06 | ≤0.015-0.03 | 0.03 | 0.03 | 0.03 |
| Burkholderia cepacia | 16 | 8 (8-50) | — | (2->256) | 4-25 | 4-256 | 12.5-256 | 4-256 | 64 |
| Campylobacter jejuni | 8 | (0.25-2) | 0.5 | 0.12-64 | 0.25-32 | 0.12-32 | 0.25-4 | 0.06-0.13 | — |
| Chlamydia pneumoniae | — | — | — | 2 | 1 | 0.5-1 | 0.12 | 0.06-1 | 0.25 |
| Chlamydia trachomatis | — | ≥16 | — | 0.5-2 | 2 (0.25-4) | 0.25-0.5 | 0.06 | 0.06 (0.015-0.12) | — |
| Citrobacter spp. | 8 | 0.5 (<0.25-50) | 0.4-1 | 0.06-0.25 | 0.25-1 | 0.13-0.5 | 0.25-2 | 0.25-2 | 2 (0.004-16) |
| Enterobacter aerogenes | 8 | 0.5 (0.2-2) | 0.25 | 0.5 (0.03->16) | 0.25 (0.1-1) | 0.5 (0.06-16) | 1 (1-16) | 2 (0.25->16) | 0.25 (0.008-2) |
| Enterobacter cloacae | 8 | 0.5 (<0.25-2) | 0.5 | 0.25 (0.025-2) | 1 (0.12-1) | 0.5 (0.05-2) | 0.5 (0.06-1) | 1 (0.06-2) | 0.25-1 (0.008-16) |
| Escherichia coli | 4 | 0.12 (0.01-0.5) | 0.12-0.25 | 0.25 (0.015->128) | 0.25 (0.06-0.25) | 0.5 (0.025-32) | 0.25 (0.016-8) | 0.25 (0.008-32) | 0.016-0.03 (0.004-32) |
| Haemophilus influenzae | 1 | 0.06 | 0.06 | ≤0.015-0.06 | 0.05-0.12 | 0.015-≤0.5 | 0.013-0.03 | 0.03-0.06 | ≤0.004-0.03 |
| Klebsiella pneumoniae | 8-16 | 0.5 (0.2-2) | 2 (0.5-2) | 0.5 (0.03->4) | 2 (0.25-2) | 0.5 (0.03->8) | 1 (0.03->4) | 1 (0.13-1) | 0.25 (0.06->4) |
| Legionella spp. | 1 | (0.2-2) | — | 0.016-0.06 | 0.03-0.12 | 0.016-0.03 | 0.03 | 0.06 | 0.003-0.03 |
| Moraxella catarrhalis | 2 | 0.4 | 0.25 | ≤0.015-0.12 | 0.06-0.12 | ≤0.03-≤0.5 | ≤0.03-0.05 | ≤0.015-0.12 | ≤0.004-0.015 |
| Morganella morganii | 8 | 0.12 (<0.06-25) | (0.25-4) | 0.06 (0.015-1) | 0.25 (0.12-0.25) | 0.12 (0.06-1) | 0.5 (0.25-2) | 0.5 (0.13-4) | 0.12 (0.016-8) |
| Mycoplasma hominis | >256 | 8-16 | 4 | 0.5-4 | 0.5 (0.25-4) | 0.25 (0.03-2) | 0.12 (0.03-0.25) | 0.06 (0.03-0.25) | 0.06 (0.015-0.12) |
| Mycoplasma pneumoniae | — | 12 | 4 | 1-5 | 1 | 0.5-2.5 | 0.13-0.5 | 0.12-0.3 | 0.25 |
| Neisseria gonorrhoeae | 1 | 0.06 | 0.06 | 0.001-2 | 0.03-2 | ≤0.008-2 | 0.004-0.025 | 0.015-1 | — |
| Neisseria meningitidis | 0.5 | 0.03 | 0.03 | 0.004-0.008 | 0.03 | ≤0.008-0.016 | ≤0.008 | ≤0.008-0.016 | 0.002 |
| Proteus mirabilis | 8 | 0.1 (0.12-0.5) | 0.25 | 0.12 (0.025-4) | 0.25 (0.12-0.5) | 0.12 (0.05-4) | 0.25 (0.2-4) | 0.5 (0.25-16) | 0.12-0.5 (0.03-8) |
| Proteus vulgaris | 8 | 0.1 (0.12-0.5) | 0.25 | 0.06 (0.03-0.12) | 0.5 (0.12-0.5) | 0.12 (0.06-0.25) | 0.39 (0.25-0.5) | 1 (0.5-1) | 0.12 (0.03-0.12) |
| Providencia rettgeri | 16 | 2 (0.25-3.1) | 0.5 | 0.5 (0.025-4) | 2-4 | 1 (0.1-4) | 0.5 | 1 (0.5-2) | — |
| Providencia stuartii | 32 | 2 (<0.25-2) | 4 | 4-8 (0.12->16) | >8 (1->16) | 4 (0.25->16) | 0.5 | 1 (0.5-2) | 0.25-16 (0.015-1) |
| Pseudomonas aeruginosa | 16 | 2 (2-16) | 2 | 0.5-128 | 4->128 | 2-128 | >4-32 | 8->128 | 4-8 (0.03-256) |
| Salmonella spp. | 2-4 | ≤0.06 (≤0.06-0.25) | 0.12 | 0.01-0.25 | 0.12-0.5 | 0.03-0.25 | 0.06-0.25 | 0.12-0.25 | 0.015-0.12 |
| Serratia marcescens | ≥100 | 1 (0.025-50) | 1 (1-8) | 2 (0.5-12.5) | 4 (1-25) | 2 (0.25-8) | 4 (2-12.5) | 4 (0.5-8) | 1-2 (0.008-4) |
| Shigella spp. | 8 | ≤0.06-0.12 | 0.25 | 0.008-≤0.06 | 0.06-0.12 | 0.016-0.03 | 0.016-0.03 | 0.03-0.06 | ≤0.015-0.25 |
| Stenotrophomonas maltophilia | 16 | 2 (2-25) | 4 | >2-32 | 8-64 | 2-32 | 2-32 | 1-128 | 4 (0.016-16) |
| Yersinia enterocolitica | 2 | ≤0.12 | 0.25 | 0.016-0.06 | 0.12-0.25 | 0.03-0.06 | 0.06 | 0.06-0.12 | 0.015-0.03 |
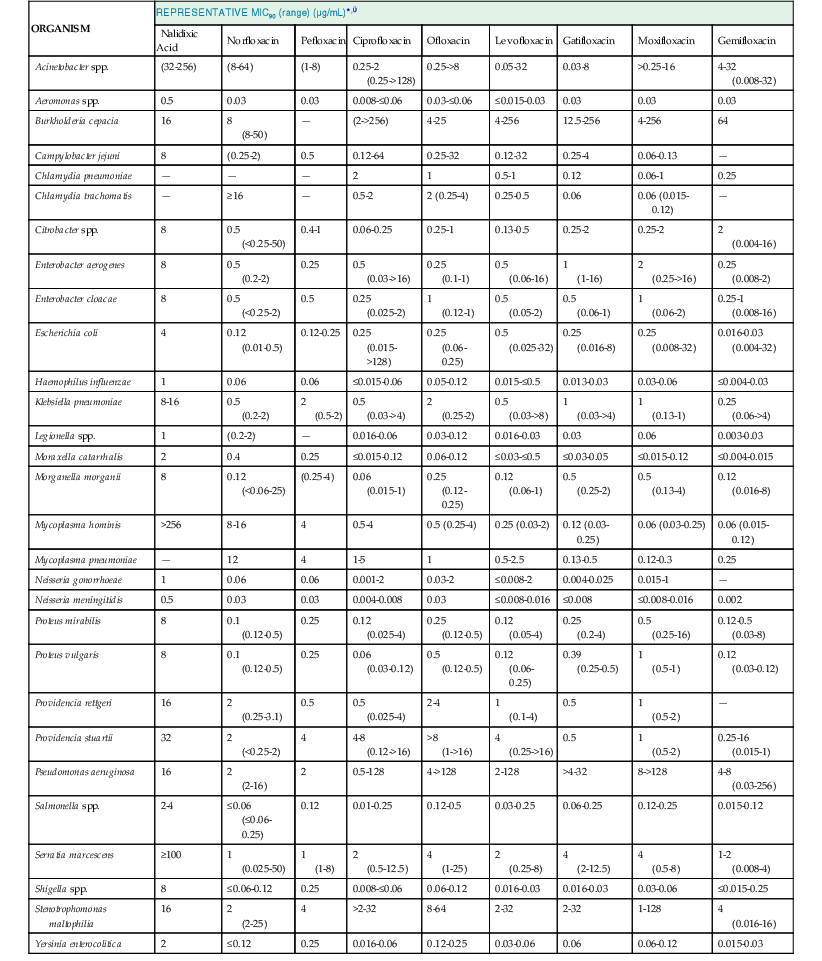
* MIC90, minimal inhibitory concentration for 90% of strains.
† Resistance has been increasing in many hospital pathogens over time. Therefore, it is important to consult individual antibiograms, because resistance can vary among institutions.
Data from Eliopoulos and Eliopoulos,73,74 Jones et al,76 García et al,80 and Bebear et al.518
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


