OUTLINE
COVID-19 Manifestations in the Respiratory System, 100
Clinical Presentation, 100
Complications, 101
Acute Respiratory Distress Syndrome, 102
COVID-19 Acute Respiratory Distress Syndrome, 111
Lung Microbiome and COVID-19, 114
Specific COVID-19 Pulmonary Complications, 115
COVID-19 Long-Term Pulmonary Complications, 116
Treatment and Management of Salient Pulmonary Manifestations and Complications of COVID-19, 118
Treatment of Hypoxemia, 118
Management of Respiratory Failure, 119
Management of COVID-19 Pneumonia and Pneumonitis, 120
Management of COVID-19 Acute Respiratory Distress Syndrome, 121
Management of Barotrauma, 122
Management of Thromboembolic Pulmonary Manifestations of COVID-19, 123
Management of Cytokine Storm and Systemic Inflammatory Response Secondary to COVID-19, 124
Current Guidelines and Medications Available for Management of COVID-19, 125
Anti-inflammatory and Antiviral Medications Used in COVID-19, 125
Antibiotic Use in COVID-19, 126
General Principles of Management of Long-Term Pulmonary Sequelae, 126
Case Reports Highlighting the Varied Pulmonary Manifestations of COVID-19, 127
Case Report 5.1 COVID-19–Associated Pneumonia and Pneumomediastinum, 127
Case Report 5.2 COVID-19–Associated Acute Respiratory Distress Syndrome, 127
Case Report 5.3 COVID-19–Associated Pulmonary Embolism, 127
Conclusion, 128
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative pathogen for coronavirus disease 2019 (COVID-19), as its name signifies, predominantly infects the lung, resulting in mild to severe lung injury. Most of the mortality and morbidity attributed to COVID-19 is a direct consequence of the lung pathological processes and the resulting complications. The cardinal clinical manifestation of COVID-19, ARDS, and in the severest form of the disease, multiorgan failure, paint a grave picture of the damage that a SARS-CoV-2 infection can do to infected individuals.
COVID-19 Manifestations in the Respiratory System
Clinical Presentation
The classic symptom complex of COVID-19 such as fever, cough, sore throat, and difficulty in breathing with underlying hypoxemia, pneumonia, and the varied manifestations in different systems of the body have been covered extensively in other chapters of this book. The common symptoms, including comorbidities and other medical conditions that can predispose SARS-CoV-2–infected individuals to severe forms of COVID-19 are summarized in Table 5.1 , which is adapted from published Centers for Disease Control and Prevention (CDC) documents. , The incubation period of COVID-19 is generally within 14 days after exposure, with most cases occurring approximately 4 to 5 days after exposure. , The pulmonary spectrum of pathological conditions ranges from mild upper respiratory tract involvement to ARDS leading to respiratory failure, with the spread of the infection to the lower respiratory tract. The range of associated symptoms of COVID-19 was illustrated in a report of 370,000 confirmed COVID-19 cases nationwide provided by the CDC. Cough was found in 50% of all reported cases with other associated symptoms, including fever (43%), myalgia (36%), headache (34%), dyspnea (29%), sore throat (20%), diarrhea (19%), nausea and vomiting (12%), and loss of smell or taste, abdominal pain, and rhinorrhea (10% each). Of all patients presenting with SARS-CoV-2 infection, 33% were asymptomatic, many of whom still had objective clinical manifestations such as ground-glass opacities (GGOs) visible on computed tomography (CT) scan and other nonspecific imaging abnormalities. The spectrum of the clinical disease varied from mild to critical, depending on age, other risk factors, and comorbidities. Almost all critically ill patients required hospitalization. In a study conducted in New York in May 2020, of 2741 critically hospitalized patients, 664 (24%) patients died or were transferred to hospice.
| Symptomatology and Comorbidities of COVID-19 | |
|---|---|
| Category | Listing |
| Symptoms | Fever, cough, shortness of breath, fatigue, body pain, headache, new loss of taste or smell, sore throat, running nose, nausea and/or vomiting, diarrhea |
| Comorbidities and medical conditions predisposing individuals to severe COVID-19 | Cancer |
| Cerebrovascular disease | |
| Chronic kidney disease | |
| Chronic lung diseases Interstitial lung disease Pulmonary embolism Pulmonary hypertension Bronchopulmonary dysplasia Bronchiectasis Chronic obstructive pulmonary disease | |
| Chronic liver diseases Cirrhosis Nonalcoholic fatty liver disease Alcoholic liver disease Autoimmune hepatitis | |
| Diabetes mellitus (types 1 and type 2) | |
| Heart conditions Heart failure Coronary artery disease Cardiomyopathies | |
| Mental health disorders Mood disorders including depression Schizophrenia spectrum disorders | |
| Obesity (BMI ≥30 kg/m 2 | |
| Pregnancy and recent pregnancy | |
| Smoking (past and present) | |
| Tuberculosis | |
Based on the Chinese Centers for Disease Control and Prevention data, 81% of patients had a mild infection, 14% had a severe infection, and 5% manifested critical illness. The criteria for severe cases were described as patients presenting with dyspnea, hypoxia, or more than 50% lung involvement on imaging within 24 to 48 hours of hospitalization; critical cases were determined based on respiratory failure and multiorgan failure. The overall case fatality reported was 2.3%.
Severe illness can occur at any age but was more prominent in advanced age and in patients with significant comorbidities. Certain demographic populations have been associated with severe illness and primarily constitute communities of color, such as African Americans, Hispanics, and South Asians. Additionally, some laboratory abnormalities have been associated with severe outcomes, as shown in Table 5.2 . A genome-wide association study raised concern for the higher risk for specific host genetic factors associated with the ABO blood group system for severe disease. Ray et al. looked at the association between ABO/Rh blood groups and susceptibility to SARS-CoV-2 infection and severe COVID-19 and reported that individuals with O and Rh-negative blood groups have a lower risk for both.
| Laboratory Abnormality | Possible Threshold | Reference Range |
|---|---|---|
| d -Dimer | 1000 ng/ml | ≤500 ng/mL |
| C-Reactive protein (CRP) | 100 mg/L | 0.0–0.9 mg/dL |
| Lactate dehydrogenase (LDH) | 245 U/L | 117–224 U/L |
| Troponin | 2 × upper limits | 0.00–0.05 ng/mL |
| Ferritin | 500 µg/L | 30–490 ng/mL |
| Creatine phosphokinase (CPK) | 2 × upper limits | 35–232 U/L |
| Absolute lymphocyte count | <800 µL | 1000–800 µL |
a Elevated laboratory biomarkers such as d -dimer, CRP, LDH, troponin, ferritin, and CPK are associated with severe COVID-19. 9
Complications
Drake et al. performed a prospective, multicenter cohort study of more than 70,000 patients admitted with COVID-19 to 302 hospitals across the United Kingdom (UK) over 7 months in 2020 and characterized the organ-specific and systemic complications of COVID-19, which are shown in Table 5.3 . Systemic, respiratory, and renal problems were the top three most frequent complications of COVID-19 among all the hospitalized patients with COVID-19 and among the patients who survived the disease; respiratory, renal, and cardiovascular complications topped the list among hospitalized patients who did not survive (see Table 5.3 ). Respiratory system complications were observed in 18% of all hospitalized patients; it was 14% among the survival group and twice that rate (28%) among the nonsurvival cohort (see Table 5.3 ). Among the various respiratory system complications presented in Table 5.4, ARDS was the most prevalent; 13% of all hospitalized patients with COVID-19 satisfied the criteria for likely ARDS, which the investigators assigned clinically or based on fulfilling one or more of the following: (1) receiving extracorporeal membrane oxygenation (ECMO), (2) nursed in a prone position and receiving mechanical ventilation, and (3) receiving mechanical ventilation with a ratio of partial pressure of arterial oxygen to fraction of inspired air of 300 mm Hg or less (PaO 2 /FiO 2 ≤300 mm Hg).
| Systemic/Organ | Systemic and Organ-Specific Complications Based on Outcomes: Survived (n = 50,105)/Died (n = 23,092)/Total (n = 73,197) | ||
|---|---|---|---|
| Survived Cohort Complications n (%) | Died Cohort Complications n (%) | Total Complications n (%) | |
| Systemic | 7423 (14.8) | 4472 (19.4) | 11,895 (16.3) |
| Respiratory system | 7028 (14.0) | 6458 (28.0) | 13,486 (18.4) |
| Renal | 10,059 (20.1) | 7693 (33.3) | 17,762 (24.3) |
| Gastrointestinal (including liver) | 4837 (9.7) | 3064 (13.3) | 7901 (10.8) |
| Cardiovascular | 4035 (8.1) | 4938 (21.4) | 8973 (12.3) |
| Neurological | 1880 (3.8) | 1235 (5.3) | 3115 (4.3) |
| Respiratory System Complications | Males (n = 41,025) n (%) | Females (n = 31,977) n (%) | Total (n = 73,002) n (%) |
|---|---|---|---|
| Cryptogenic organizing pneumonia | 50 (0.1) | 42 (0.1) | 92 (0.1) |
| Likely acute respiratory distress syndrome | 6417 (15.6) | 3251 (10.2) | 9668 (13.2) |
| Pneumothorax | 466 (1.1) | 227 (0.7) | 693 (0.9) |
| Pleural effusion | 2583 (6.3) | 1940 (6.1) | 4523 (6.2) |
| Likely bacterial pneumonia | 260 (0.6) | 99 (0.3) | 359 (0.5) |
| Pulmonary embolism | 622 (1.5) | 382 (1.2) | 1004 (1.4) |
After presentation of the basic clinical picture of COVID-19, including symptomatology and potential complications, we provide a detailed description of ARDS, its epidemiology, pathogenesis, clinical presentation, and management as a foundation for understanding the pulmonary pathological processes and specific complications of COVID-19.
Acute Respiratory Distress Syndrome
The ARDS syndromic complex is characterized by pulmonary edema, ventilation-perfusion (V/Q) defects, breathlessness, hypoxemia, and significant alveolar damage. The original description of the syndrome goes back more than 50 years to 1967. A consensus definition of ARDS in adults was proposed in 2012 and referred to as the Berlin definition ; it was modified in 2016 and called the Kigali modification. The definitions are based on four parameters as depicted in Table 5.5 . ARDS, briefly, is hypoxemia with a partial pressure of arterial oxygen (PaO 2 ) to fraction of inspired oxygen (FiO 2 ) ratio of 300 mm Hg or less and presence of bilateral lung infiltrates seen on chest imaging (x-ray, CT), in the absence of supporting evidence for left heart failure.
| Berlin Definition | |
|---|---|
| Characteristics | Description |
| Timing of respiratory failure | Within 1 wk of a known clinical insult or new or worsening respiratory symptoms |
| Origin of pulmonary edema | Respiratory failure not fully explained by cardiac failure or fluid overload Need objective assessment, such as echocardiography, to exclude hydrostatic edema if no risk factor is present |
| Chest imaging | Bilateral opacities on chest radiograph or chest computed tomography not fully explained by effusion, collapse, or nodules |
| Oxygenation | Acute onset of hypoxemia defined as PaO 2 /FiO 2 ≤300 mm Hg on at least PEEP 5 cm H 2 O |
| Mild | PaO 2 /FiO 2 201–300 mm Hg |
| Moderate | PaO 2 /FiO 2 101–200 mm Hg |
| Severe | PaO 2 /FiO 2 ≤100 mm Hg |
| Kigali Modification | |
| Timing and origin | Same as the Berlin criteria given above |
| Chest imaging | Bilateral opacities on chest radiograph or chest ultrasound not fully explained by effusion, collapse, or nodules |
| Oxygenation | SpO 2 /FiO 2 ≤315 and no PEEP requirement |
Epidemiology of Acute Respiratory Distress Syndrome
One of the largest pre-COVID ARDS studies, the LUNG-SAFE study, conducted in 2014 assessed an in-hospital mortality of 40% with a range of 35% to 46% based on severity of ARDS at the time of diagnosis. , The study estimated the prevalence of ARDS among patients hospitalized in intensive care units (ICUs) based on data from 50 countries to be 10.4% and also concluding that ARDS appeared to be underrecognized, undertreated, and underreported. In the United States the annual incidence of ARDS was estimated to be 190,000 cases, with a reported mortality of 38.5%. Several comorbidities, including alcohol abuse, cigarette smoking, polluted air, and hypoalbuminemia, have been recognized as contributing to the severity of the syndrome. There are also significant racial, ethnic, and sex disparities, as well as differences in susceptibility based on population density and socioeconomic status for ARDS morbidity and mortality, with higher rates among the Black and Hispanic populace, males, low-income groups, and people living in high-density population regions. , ,
Cause of Acute Respiratory Distress Syndrome
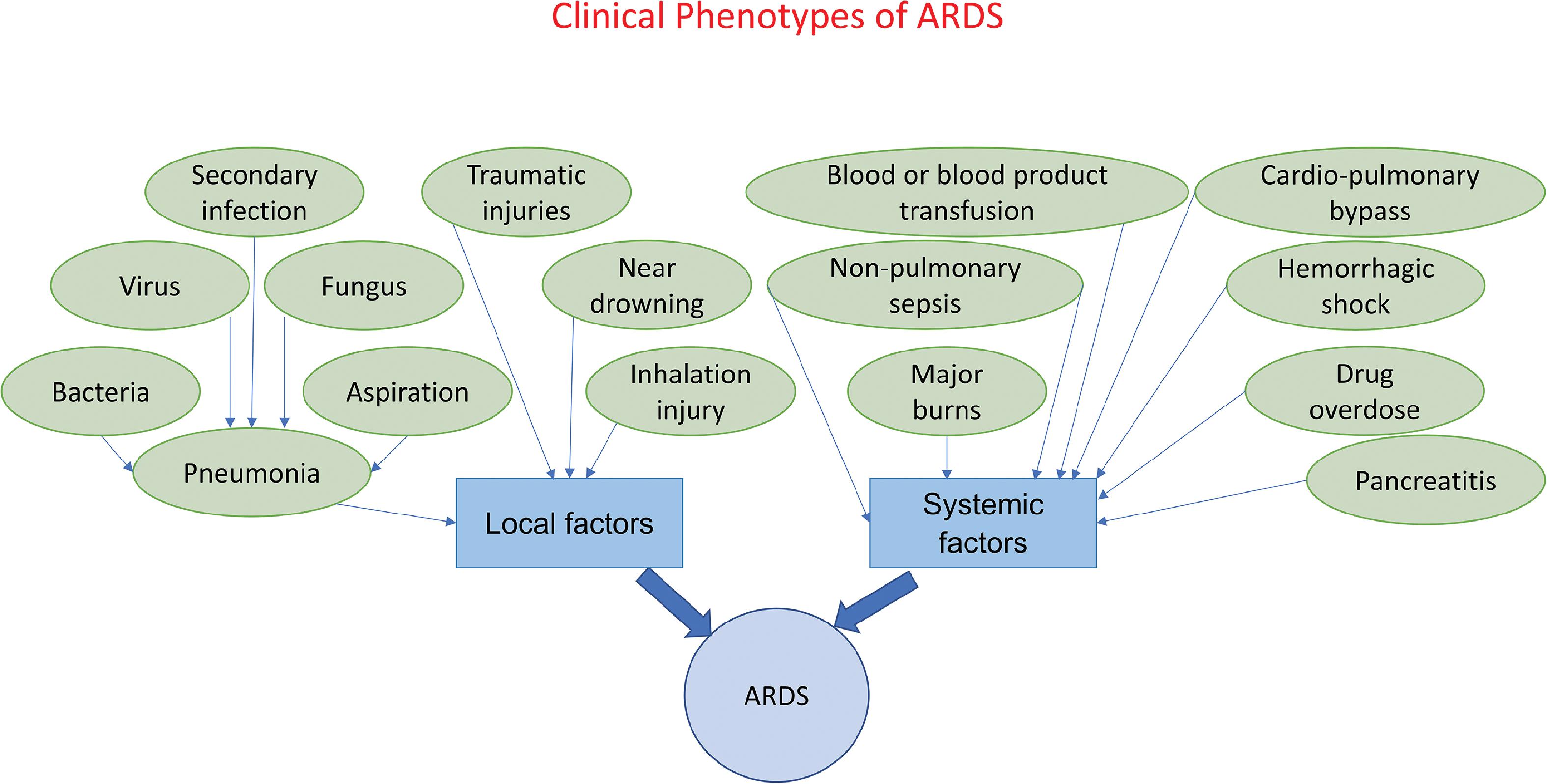
The causative risk factors for ARDS, which generally occurs in the setting of pneumonia can be broadly categorized as local and systemic. Respiratory bacterial, viral, fungal, and secondary infections that cause pneumonia, aspiration pneumonia, lung trauma resulting from blunt or penetrating injuries, and inhalation injuries are common local causes; systemic causes include nonpulmonary sepsis, acute pancreatitis, blood or blood product transfusion, drug overdose, major burns, cardiopulmonary bypass, graft-versus-host disease, and high-altitude pulmonary edema (HAPE). ,
Pathophysiology of Acute Respiratory Distress Syndrome
Histopathology With Alveolar Focus
The characteristic pathological hallmark of ARDS is diffuse alveolar damage which compromises the basic lung physiology of ventilation and perfusion, leading to hypoxemia. , , The temporal evolution of the alveolar damage occurs over weeks in three phases. The first or the exudative phase starts in the first week with alveolar epithelial injury affecting both type I and type II pneumocytes. This causes denudation of the alveolar membrane and increased permeability. Eosinophilic deposits result in hyaline membrane formation along the denuded alveolar basement membrane, a classic finding in diffuse alveolar damage along with white blood cell agglutination and formation of platelet-fibrin thrombi. Alveolar fluid reabsorption by ion transport, a normal function of alveolar cells, is affected resulting in accumulation of exudative fluid in the alveolar cavity. Macrophages residing in the alveolus secrete proinflammatory factors such as interleukin-8 (IL-8) and monocyte chemoattractant protein-1, which attract neutrophils and other macrophages into the lung, aggravating the lung injury. Concomitant endothelial damage to the capillaries lining the alveolus results in the disruption of endothelial barrier leading to interstitial flooding, with the excess fluid finding its way to the alveolar space and causing intraalveolar flooding and exacerbation of edema. The nature of insult to the endothelial cells is not well understood but apoptosis and pyroptosis have been proposed as plausible mechanisms. , Endothelial cell activation results in release of angiopoietin-2, leading to formation of neutrophil-platelet aggregates that pass through the weakened endothelial barrier and infiltrate the alveolar space. The neutrophil-platelet aggregates manifest thromboinflammatory mechanisms such as neutrophil extracellular traps (NETs), which are implicated in both capillary endothelial and alveolar epithelial barrier perturbations. , Other causative factors responsible for endothelial wall damage are pathogens and circulating toxins, alveolar macrophage factors, and proinflammatory cytokines and chemokines. , The cascade of events set in motion by epithelial and endothelial injury causing alveolar flooding with exudate results in surfactant dysfunction, which promotes atelectasis and biophysical injury to the lung.
The second phase, called the proliferative phase, sets in motion the repair mechanisms to tackle the intraalveolar and interstitial exudative edema of phase 1. Beginning in the second week, the proliferative phase lasts for about 2 weeks. The key events of this phase include interstitial fibroblast proliferation with secretion of epithelial growth factors, alveolar type II cell hyperplasia with differentiation into alveolar type I cells, phagocytosis of apoptotic neutrophils, and reestablishment of junctions between epithelial cells with restoration of the integrity of the epithelial barrier. Epithelial barrier repair enables reabsorption of edematous fluid into the interstitial space. Fibroblasts transform the exudate to cellular granulation tissue with matrix morphology, which eventually turns into fibrous tissue with the deposition of collagen. Likewise, in the lung capillaries endothelial cell proliferation sets in with repair of endothelial disruption and restoration of the endothelial barrier function. The pathogenetic mechanisms of the three phases of ARDS are enumerated in Figs. 5.1 and 5.2 , which illustrate the pathological process of the injured alveolus in the exudative phase and the repair mechanisms of the proliferative phase with the healthy alveolus as reference.
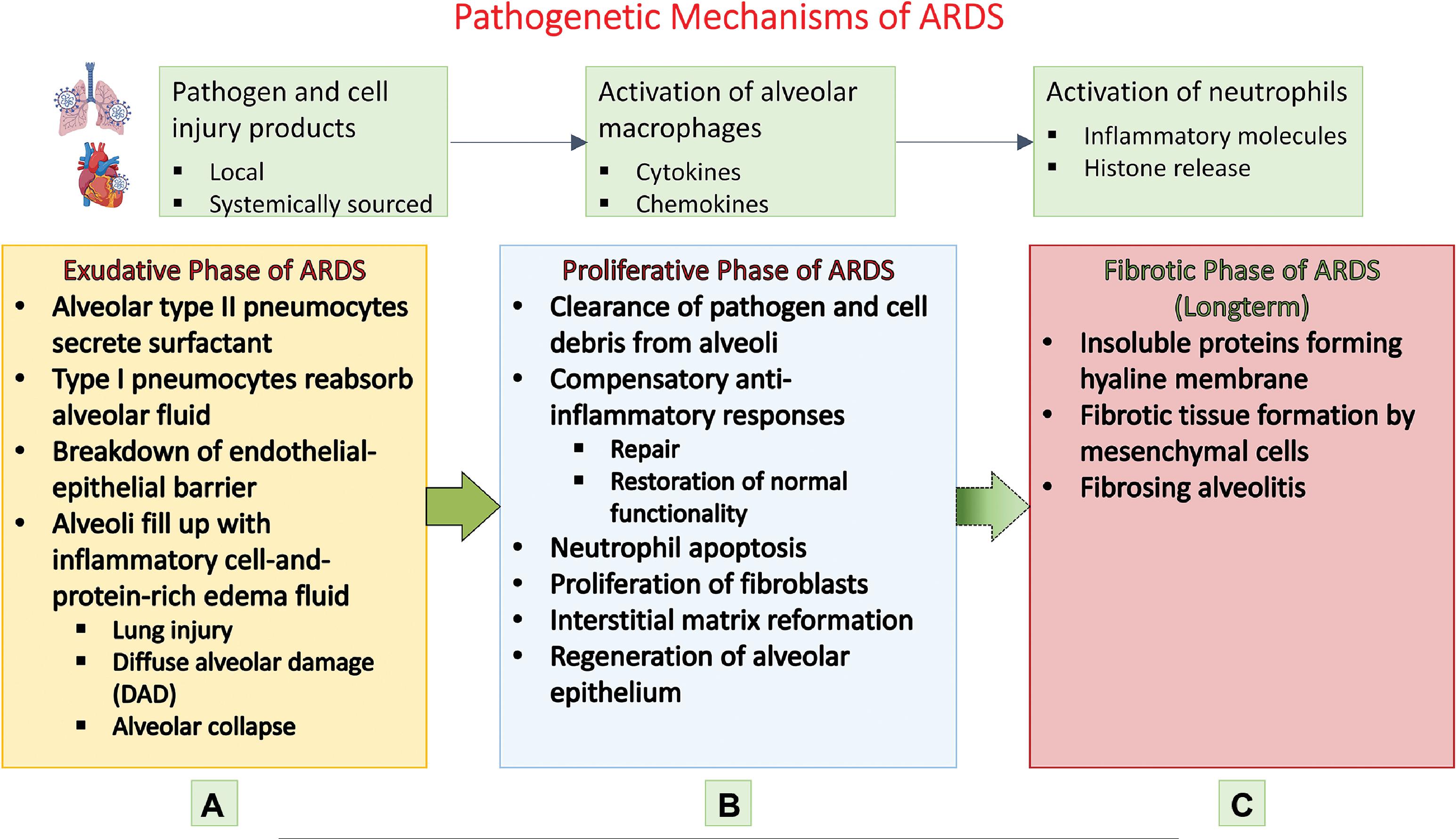
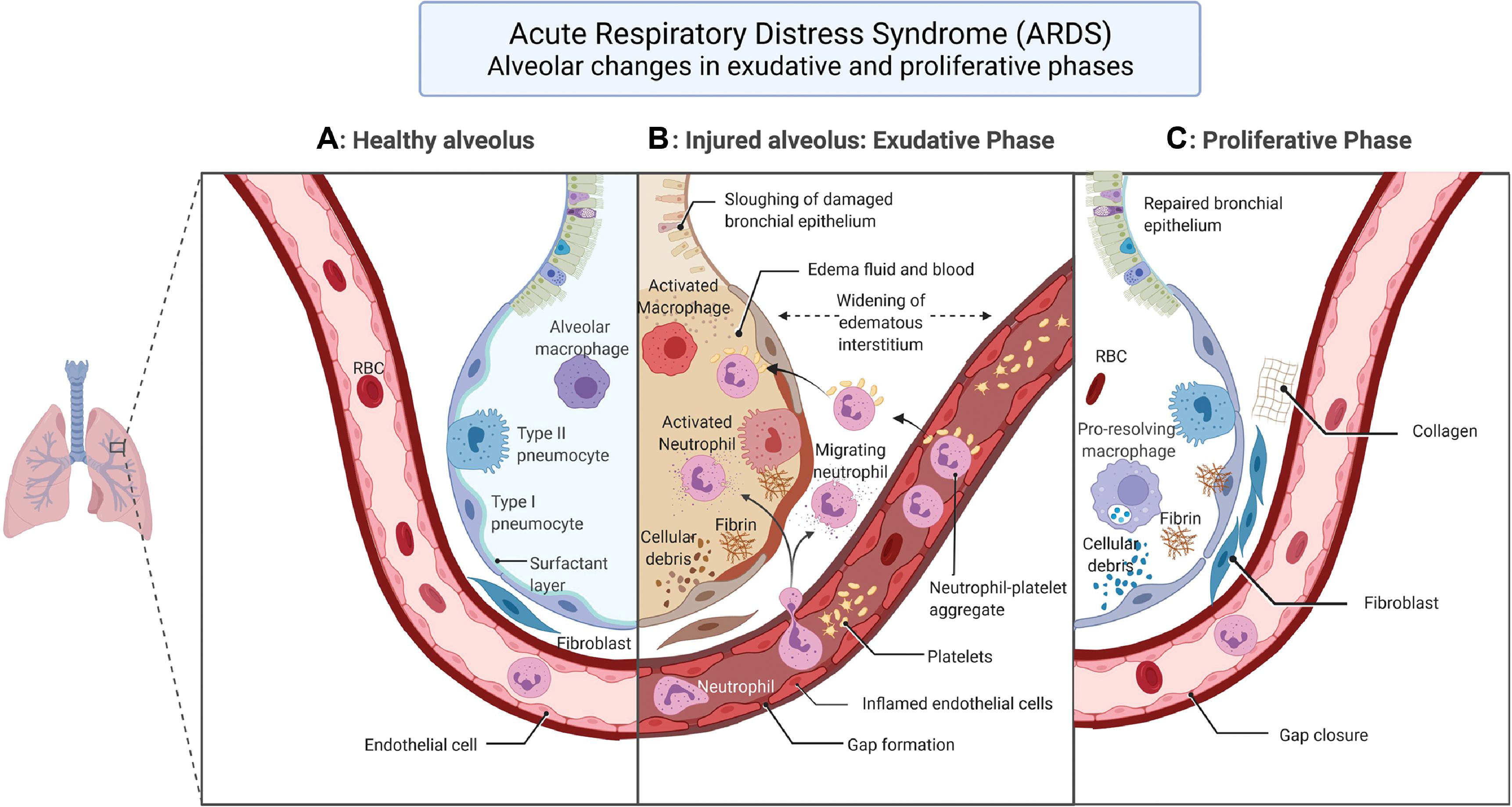
The third or fibrotic phase necessitates mechanical ventilation and is typically seen in patients surviving beyond 3 to 4 weeks from the onset of ARDS. This phase is marked by significant interstitial and intraalveolar fibrosis with lung remodeling by thick collagenous connective tissue. , The process of fibrosis acts as a nonspecific response to the lung injury and resembles the lung tissue reorganization that occurs in typical organizing pneumonia.
Additional pathological features of ARDS are summarized in Table 5.6 and salient histopathological changes observed in the temporal evolution of diffuse alveolar damage are shown in Table 5.7 . After exploration of the pathophysiology of ARDS, the chapter discusses alterations in respiratory mechanics. To formulate a rational approach to the management of ARDS it is necessary to understand the changes in respiratory mechanics in ARDS.
| ARDS | |
|---|---|
| Pathophysiological Characteristics | Description |
| Diffuse alveolar damage (DAD) | The predominant pathological condition is epithelial injury that involves types I and II pneumocytes and dissociation of junctions between these alveolar cells resulting in increased permeability. Neutrophil migration resulting from endothelial injury leads to alveolar epithelial injury by the perturbation of intercellular junctions and causing enhanced epithelial permeability. See Table 5.7 for a list of additional histopathological details of DAD and its three phases. |
| Impaired alveolar fluid clearance | Alveolar fluid reabsorption is performed by types I and II alveolar pneumocytes by active ion transport into the interstitium from which clearance occurs by lymphatic channels. Because of alveolar cell damage, permeability is lost, affecting ion transport resulting in accumulation of exudative fluid in alveolar spaces. |
| Ventilation-perfusion abnormalities (gas exchange anomalies) | Type II alveolar cells secrete surfactant, which plays a key role in gas exchange in the lungs by keeping the alveoli open based on its capacity to lower surface tension. The loss of surfactant causes ventilation-perfusion mismatch and impairs carbon dioxide elimination. |
| Reduction in lung compliance | Increased permeability, loss of surfactant resulting from damage to type II pneumocytes and accumulation of fluid in the alveolus leads to reduction in lung volume available for gas exchange. This in turn causes increased dead space in the lungs and lowering of lung compliance. |
| Increased pulmonary vascular resistance | Impaired carbon dioxide elimination causes hypercapnia leading to pulmonary vasoconstriction. This, along with thromboembolism and interstitial edema, results in increased pulmonary vascular resistance and pulmonary hypertension. |
| Endothelial injury | Endothelial cell damage results in leakage of fluid across the capillary barrier into the interstitial space. This results in an edematous interstitium with fluid moving into the alveoli through the damaged epithelial barrier. |
| Thromboembolism | Postmortem studies of lungs from patients dying of ARDS have consistently demonstrated thromboembolic lesions. Macrothrombi and microthrombi have been extensively observed in arterioles and capillaries of the lung. Two types of microthrombi have been reported: (1) composed of hyaline platelet-fibrin seen in capillaries and arterioles and (2) laminated fibrin clots visualized in preacinar and intraacinar arteries. |
| Ventilatory support injury | See Table 5.8 |
| ARDS DAD | ||
|---|---|---|
| Exudative Phase (wk 1) | Proliferative Phase (wk 2 and 3) | Fibrotic Phase (>3 wk) |
| Interstitial edema Alveolar edema Leukocyte infiltration and agglutination Pneumocyte type I necrosis Endothelial cell necrosis Alveolar basement membrane denudation Hyaline membrane formation Platelet-fibrin thrombi | Interstitial fibroblast proliferation Pneumocyte type II proliferation and differentiation into pneumocyte type I Reestablishment of tight junctions and adherent junctions Restoration of epithelial barrier function Parenchymal necrosis Phagocytosis of apoptotic neutrophils Endothelial cell proliferation Restoration of endothelial barrier function Obliterative endarteritis Macrothrombi | Prolonged proliferation of fibroblasts Collagenous fibrosis Microcystic honeycombing Traction bronchiectasis Obliteration of microcapillaries Arterial tortuosity with mural fibrosis and medial hypertrophy |
Respiratory Physiology and ARDS
The honeycomb-like structure of the lung composed of a multitude of small air sacs or alveoli, capillaries, and interstitial tissue confers the lung parenchyma its elastance and compliance, enabling it to expand without ballooning during inspiration and contract without collapsing during expiration. As diffuse alveolar damage sets in, the increased permeability across the alveolar membrane and vascular endothelium results in alveolar flooding. Alveolar fluid accumulation combined with loss of surfactant results in alveoli becoming unrecruitable, leading to shrinkage of lung volume available for gas exchange and loss of compliance. This alveolar derecruitment, that is, exclusion from gas exchange, causes a physiological division of the lung into nonaerated diseased regions and aerated healthy regions. Because the number of recruitable lung units, that is, aerated healthy regions, are reduced in toto, the phenomenon has been labeled “baby lung.”
The two key blood gas anomalies of ARDS are hypoxemia and hypercapnia. The basic respiratory physiological measure that modulates these two blood gas values is the alveolar V/Q ratio, denoted by V A /Q, where V A refers to alveolar ventilation and Q is perfusion. For normal gas exchange to occur, an optimal V A /Q is needed, which is disrupted in ARDS because of the V/Q mismatch, resulting in some regions of the lung with low V A /Q ratios and other regions with high V A /Q ratios. In regions with low V A /Q ratios, perfusion is much higher with respect to ventilation, leading to hypoxemia; in regions where the V A /Q ratios are higher, ventilation is markedly more compared with perfusion, resulting in hypercapnia. Low V A /Q ratios cause shunting, the gas partial pressures reflecting those of the mixed venous blood, whereas high V A /Q ratios result in dead spaces with gas partial pressures similar to those of inspired air. In short, hypoxemia of ARDS results from blood flow to alveoli that are not ventilated and hypercapnia sets in when alveoli have proper ventilation but lack perfusion.
Lung Injury From Mechanical Ventilation
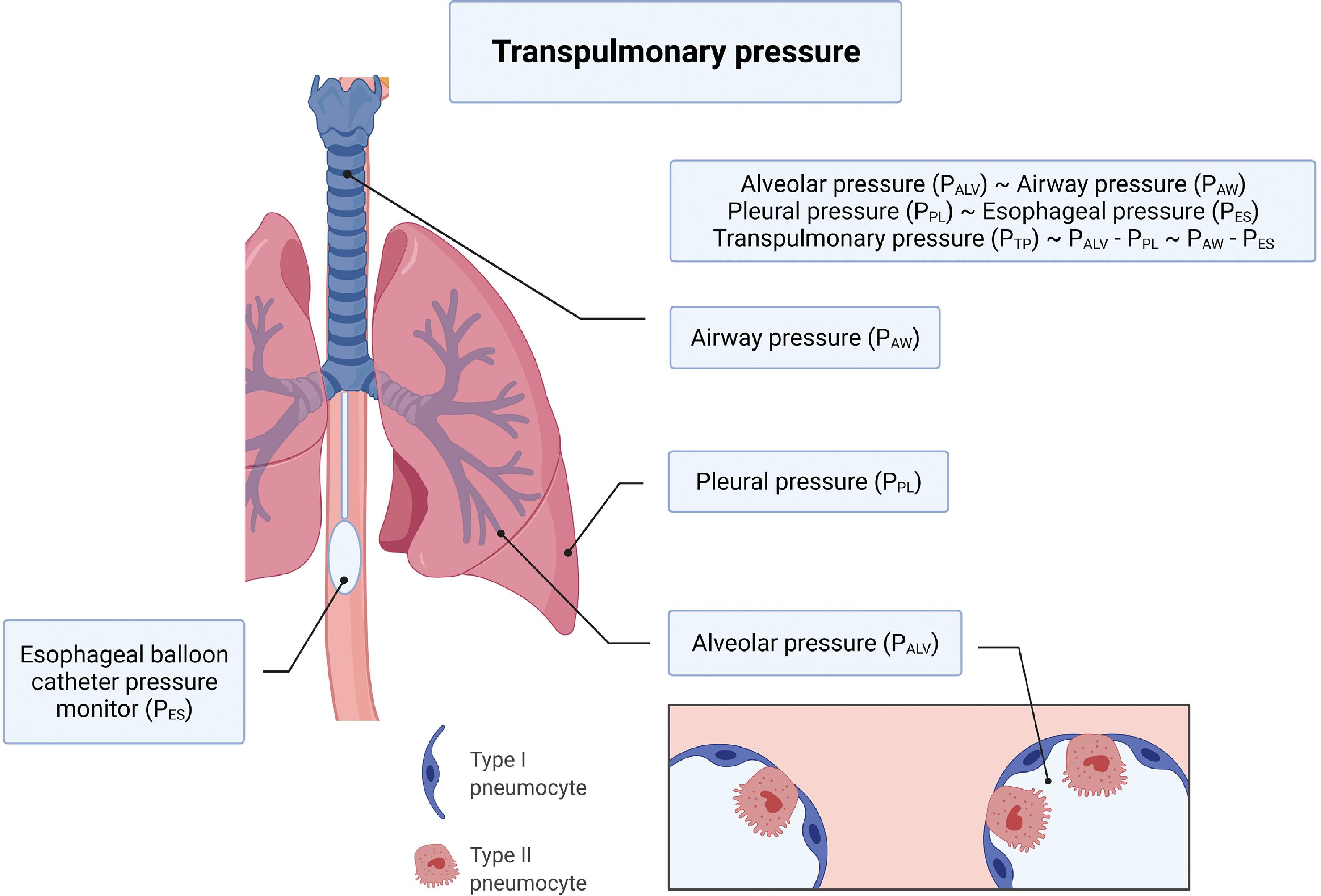
Ventilator-induced lung injury (VILI) has garnered a lot of attention over the last decade , and continues to do so with the advent of the COVID-19 pandemic. In this section we will introduce the salient types of VILI and examine how mechanical ventilation exacerbates evolving lung injury of ARDS. At the end of inspiration when there is no air flowing into the lungs, lung inflation is maintained by what is referred to as the transpulmonary pressure, which is the difference between alveolar and pleural pressures and is closely linked with VILI. Details of transpulmonary pressure and how it can be estimated in the clinical setting are illustrated in Fig. 5.3 . Lung volume and transpulmonary pressure are interrelated, and the type of injury resulting from mechanical ventilation is based on whether ventilation occurs at high or low lung volumes. , Four types of VILI have been described—barotrauma, volutrauma, atelectrauma, and biotrauma. VILI is also related to lung injury that results from the efforts to breathe spontaneously on the part of the patient during mechanical ventilation and is described as patient self-inflicted lung injury (P-SILI). , Both VILI and P-SILI are “ventilation”-induced lung injury. Table 5.8 and Fig. 5.4 summarize the types of VILI, the mechanisms underlying the injury, and their clinical manifestations.
| Mechanism of Injury | Type of Injury | Manifestations |
|---|---|---|
| Mechanical ventilation at high lung volumes Overdistension of the alveoli Repetitive strain | Barotrauma (air leaks) Volutrauma | Pneumothorax Pneumomediastinum Air embolism Subcutaneous emphysema Pulmonary edema |
| MV at low lung volumes Lung injury from opening and closing of alveoli Parenchymal shear injury Aggravated by heterogeneity | Atelectrauma | |
| Patient self-inflicted lung injury (P-SILI), that is, spontaneous breathing during MV causing lung injury High tidal volumes from effort High transpulmonary pressure from vigorous efforts (pendelluft effect) Negative pleural pressure leading to increased transvascular pressure resulting in alveoli filling up with edematous fluid Asynchrony between spontaneous effort and MV Forced expiration causes diaphragmatic shift leading to lower end-expiratory lung volume and hypoxemia | Barotrauma | Pneumothorax |
| Downstream biological effects of all types of lung injury High lung volume Low lung volume Molecular and cellular events of lung parenchymal stress Increased alveolar-capillary permeability | Biotrauma | Pulmonary edema Pulmonary fibrosis (long-term) Multiple-organ failure from proinflammatory mediators entering systemic circulation |
a The table also includes patient self-inflicted lung injury (P-SILI). Both VILI and P-SILI are “ventilation”-induced lung injury.
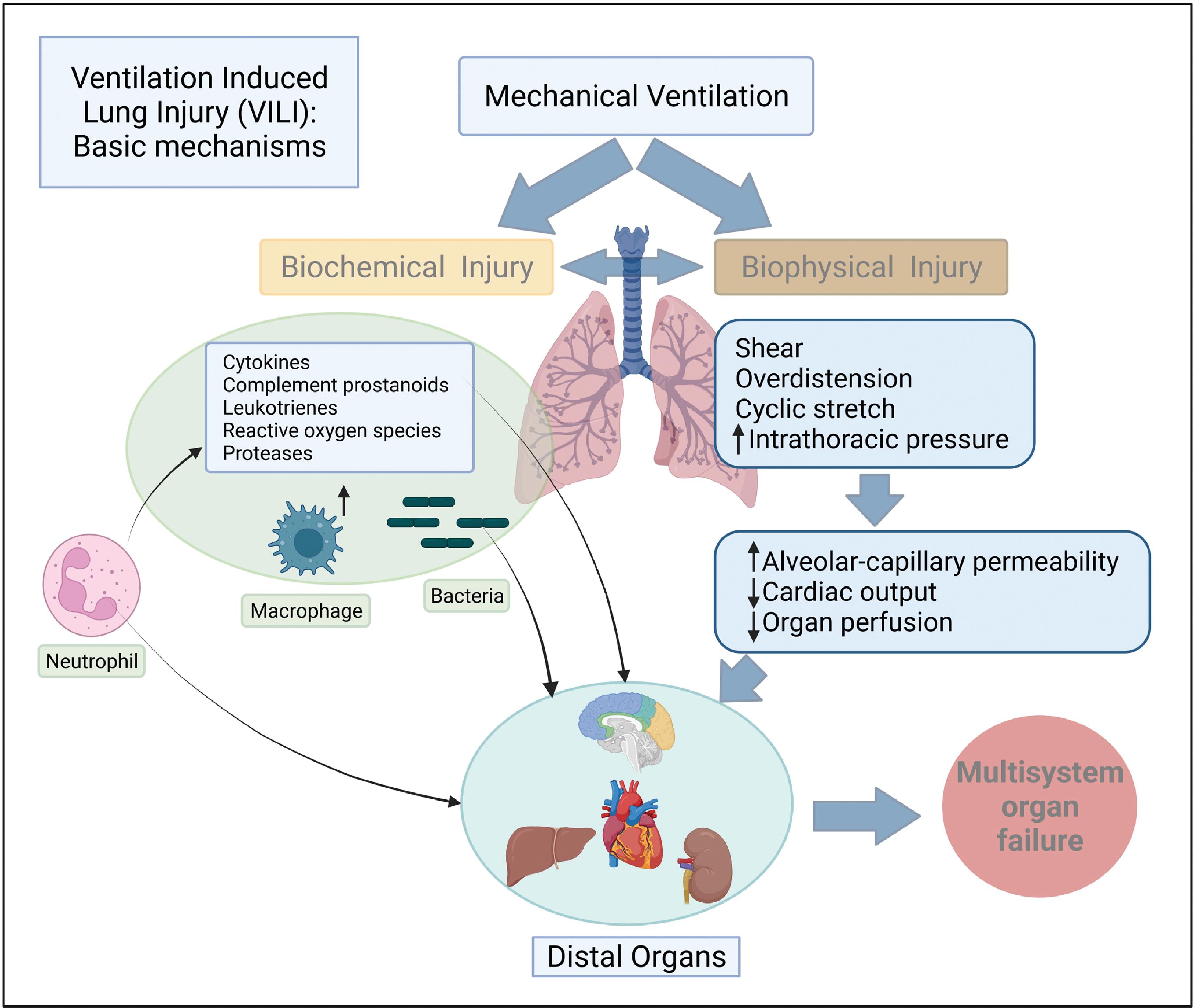
Following up on the observation that VILI originates in isolated and scattered regions of the lung parenchyma based on heterogeneity, Gaver III et al. proposed a permeability-originated obstruction response framework with four features: (1) enhanced endothelial permeability, (2) alveolar surfactant deactivation, (3) alveolar and interstitial edema, and (4) repetitive recruitment/derecruitment resulting in decreased compliance leading to atelectrauma, which has implications for mechanical ventilation management. Unless effective interventions are promptly undertaken, the proponents of the model warn that a repetitive cycle of these four steps will induce a vicious cycle resulting in increasing VILI.
Clinical Features and Diagnosis of Acute Respiratory Distress Syndrome
ARDS is characterized by the triad of rapid-onset hypoxemia, bilateral lung infiltrates, and pulmonary edema not attributable to a pathological cardiac condition. As the syndromic nomenclature based on clinical and pathophysiological criteria signifies, there is considerable heterogeneity in terms of cause, comorbidities, pathophysiology, and biology of ARDS. There is no specific biomarker to diagnose ARDS; moreover, no set of findings by themselves—molecular, clinical, radiological, or pathological—can be considered a sufficient set of markers for the diagnosis of ARDS. Thus there is a need for consensus diagnostic criteria for uniformity in approach to the diagnosis of ARDS.
ARDS diagnosis follows the Berlin definition criteria presented earlier in Table 5.1 . To recapitulate, the clinical recognition is based on (1) the onset of acute respiratory failure within 1 week of clinical insult, (2) noncardiac origin of respiratory failure, (3) bilateral opacities on chest radiograph or chest CT, and (4) hypoxemia. Although the diffuse alveolar damage feature is implicitly incorporated into the definition, autopsy studies of the lung identified diffuse alveolar damage features in only half of the patients dying from the syndrome. , Likewise, lung biopsy samples of patients hospitalized with ARDS demonstrated histological evidence of diffuse alveolar damage in similar proportions. , ,
Clinically, ARDS can be broadly categorized into two clinical phenotypes based on local or systemic etiological factors. See Fig. 5.5 for an illustration of these clinical phenotypes and their risk/causative factors. It has been shown that the risk for developing ARDS increases when the insult is local when compared with systemic factors.
Morphological phenotypes have been postulated based on radiological patterns observed in chest CT of patients with ARDS. Two distinct subgroups have been identified: (1) focal ARDS evidenced by consolidations in the lower lobes and dependent dorsal regions and (2) nonfocal ARDS with diffuse patchy opacities; the two groups had significant differences in lung physiological processes, with the lungs of the nonfocal ARDS group more recruitable than the focal ARDS group. , , Researchers have developed a noninvasive method to estimate the extent of pulmonary edema based on chest x-rays. The radiographic assessment of lung edema (RALE) score, which evaluates the extent and density of alveolar opacities on chest radiographs, was found to be highly correlated with the severity of ARDS and increased mortality from ARDS. However, the relationship of the RALE score to the focal and nonfocal ARDS groups based on chest CT findings has not been established.
Investigators have also explored protein biomarkers to stratify and discover ARDS phenotypes. In particular, Forel et al. identified a specific protein biomarker, procollagen peptide (PCP-III), with the best cutoff value from bronchoalveolar lavage fluid in persistent ARDS to identify patients with lung fibroproliferation who could likely benefit from the administration of corticosteroids. Hamon et al. used the threshold level of PCP-III greater than 9 µG/L, indicating lung fibroproliferation, and compared chest scans of 228 patients with ARDS who had a bronchoalveolar lavage fluid PCP-III level and a chest CT scan; the study demonstrated significant association between high levels of PCP-III and CT scan fibrosis scores.
Using data from two randomized controlled trials (RCTs), the ARMA and ALVEOLI, and applying a latent class analytics approach, Calfee et al. identified two subphenotypes: the hyperinflammatory, characterized by severe inflammation, shock, and metabolic acidosis, leading to worse outcomes and labeled subphenotype 2, and the less inflammatory type, denoted subphenotype 1; the distribution was 30% for subphenotype 2 and 70% for subphenotype 1. The hyperinflammatory subphenotype 2 had higher plasma levels of inflammatory biomarkers, greater vasopressor use, lower sodium bicarbonate levels, and a higher prevalence of sepsis compared with subphenotype 1; subphenotype 2 also had a differential response to positive end-expiratory pressure (PEEP).
Using a different ARDS cohort from another trial, the Fluid and Catheter Treatment Trial (FACTT), Famous et al. identified the two subphenotypes, hyperinflammatory subphenotype 2 and the less inflammatory subphenotype 1, based on levels of IL-8, bicarbonate, and tumor necrosis factor receptor-1, and reported that fluid management protocols had significantly different effects on 90-day mortality outcomes in the two subphenotypes. A fluid conservative strategy resulted in lower 90-day mortality in subphenotype 1, whereas the same approach caused an increase in 90-day mortality in subphenotype 2.
There is currently no clinically valid biological marker for detecting ARDS with high sensitivity and specificity. However, markers of epithelial and endothelial injuries have been shown to be elevated in ARDS and they could be useful in distinguishing ARDS from local and systemic insults. Plasma biomarkers of epithelial injury such as surfactant protein-D and soluble receptor for advanced glycation end products are found to be elevated in ARDS from local causes. Likewise, plasma biomarkers of endothelial injury, for example, angiopoietin-2 and von Willebrand factor antigen, are higher in ARDS from systemic causes such as nonpulmonary sepsis.
Because bacterial and viral infections are common causes, a typical ARDS workup involves blood culture, sputum or tracheal aspirate Gram staining, and culture. However, identification of the causative pathogen may not be straightforward. Nucleic acid detection methods might help improve detection and can help with the identification of both bacterial and viral pathogens. ,
Conditions that can mimic ARDS include congestive heart failure, interstitial lung diseases, connective tissue disorders such as polymyositis, vasculitis, lymphomas, and endobronchial tuberculosis; these are commonly referred to as ARDS mimics and would require additional investigations for a precise diagnosis.
Treatment and Prevention
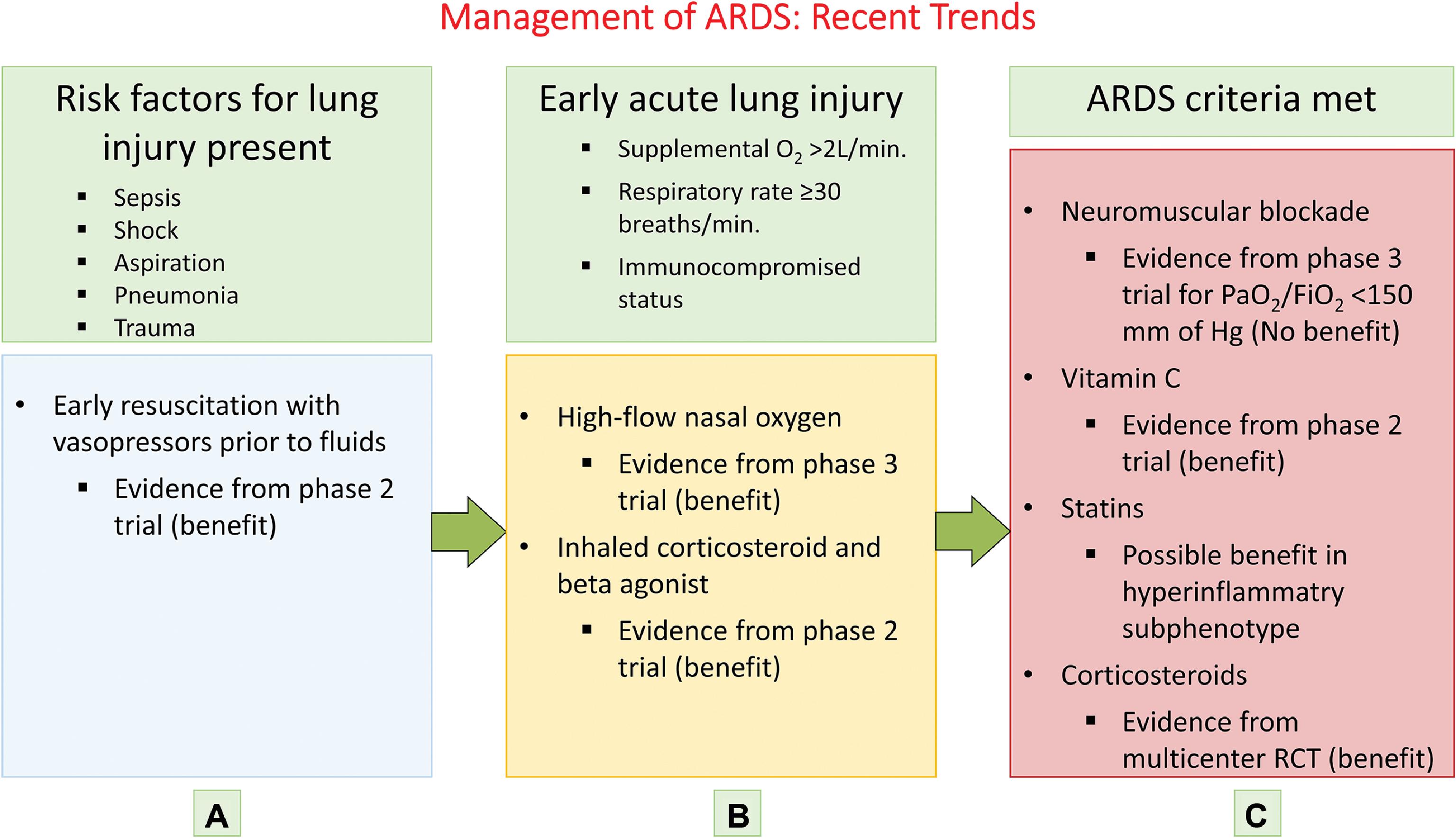
The treatment for ARDS is evolving, but it is still mostly preventive and supportive. Once the patient has transitioned from lung injury to the stage of ARDS, effective treatments are still in experimental stages. Hence the focus of investigators has broadened to include identification and treatment of patients who are at risk for developing ARDS and show early evidence of lung injury. Based on this approach, three categories have been developed: (1) patients with risk factors for lung injury such as sepsis, shock, or pneumonia; (2) patients in the early stage of acute lung injury; and (3) patients meeting ARDS criteria. These categories and modified approaches for treating them are presented in Fig. 5.6 . Table 5.9 provides the protective ventilation and intervention strategies and their rationale. ,
| ARDS: Mechanical Ventilation Guideline Recommendations | ||||
|---|---|---|---|---|
| Intervention | Severity of ARDS | Quality of Evidence | Strength of Recommendation | Recommendation Type (For/Against) |
| Mechanical ventilation using lower tidal volumes 4–8 mL/kg PBW Plateau pressure <30 cm H 2 O | Mild, moderate, and severe | Moderate | Strong | For |
| Prone positioning >12 hr/day | Severe | Moderate | Strong | For |
| Routine use of high-frequency oscillatory ventilation | Moderate and severe | High | Strong | Against |
| Higher positive end-expiratory pressure (T) | Moderate and severe | Moderate | Conditional | For |
| Recruitment maneuvers | Moderate and severe | Low | Conditional | For |
| Extracorporeal membrane oxygenation (ECMO) | Severe | Not applicable | Not applicable | More evidence needed for recommendation |
To summarize, the various strategies to reduce VILI include (1) low tidal volume ventilation to limit overdistention, (2) moderate to high PEEP to prevent derecruitment and injury from low lung volume, and (3) recruitment maneuvers with application of greater than 35 cm H 2 O airway pressure to open collapsed lung segments.
There has been a recent trend toward finding subphenotypes of ARDS with the goal of personalization of treatment based on cause, various types of biomarkers, or gene expression data. , , Sinha et al. proposed a method of clinical stratification of ARDS for subphenotype identification based on a machine learning method called latent class analysis using clinically available data. Once the model is validated using clinical data from different centers, it is possible that the machine learning model could be useful at the bedside for phenotype identification. Wick et al. discuss the promises and challenges of developing a personalized medicine approach for the management of ARDS. The review specifically identified the heterogeneity in the presentation and manifestation of ARDS and the differences in the cause of ARDS in developed and developing countries as two significant challenges for developing a patient-specific paradigm of treatment.
As Kotas and Thompson have nicely put it, ARDS is a syndrome and not a disease. To maximize desirable outcomes, it is important to identify the cause of ARDS in a patient-specific manner while following the general principles and guidelines for the management of ARDS.
Long-Term Outcomes
Patients recovering from ARDS suffer from significant long-term health consequences. Muscular weakness, anxiety, depression, and posttraumatic stress disorder (PTSD) have been reported in survivors.
With this basic layout of the different facets of ARDS, which hopefully will serve as a foundation for understanding the lung pathological processes and management of COVID-19 respiratory manifestations, the chapter will proceed to discussion of the pathophysiology, management, and complications of lung injury resulting from SARS-CoV-2 infection. The focus will be on the pathophysiology of COVID-19 lung injury, similarities and differences between COVID-19 ARDS and classic ARDS, pulmonary complications of COVID-19, management protocols, and long-term sequelae after SARS-CoV-2 infection.
COVID-19 Acute Respiratory Distress Syndrome
Epidemiology of COVID-19 Acute Respiratory Distress Syndrome
The epidemiology of SARS-CoV-2 and COVID-19 has been covered in detail in an earlier chapter. As mentioned earlier, the annual incidence of ARDS in the United States before the advent of the COVID-19 pandemic was 190,000. Based on the mortality figure of 650,000 in the United States from March 2020 to September 2021 and assuming that 80% had severe pneumonia/ARDS and attributing a mortality of 20% from COVID-19 ARDS, Wick et al. estimate an incidence of 2.5 million cases of COVID-19–associated ARDS over 18 months or about 1.7 million over a period of 12 months.
Pathophysiology of COVID-19 Acute Respiratory Distress Syndrome
In the early months of the SARS-CoV-2 pandemic, a controversy emerged on whether the pathophysiological mechanisms of COVID-19 ARDS and classic ARDS were significantly different to warrant an alternative approach to the management of the condition. However, a consensus later emerged signifying that there are more similarities than differences between COVID-19 ARDS and ARDS from other causes and that COVID-19–related lung injury should be managed based on the lung-protective strategies recommended for classic ARDS. ,
Hypoxemia and COVID-19 Lung Injury
The key manifestation in COVID-19 lung injury is hypoxemia. COVID-19 patients presenting with “silent hypoxemia,” that is, hypoxemia measured by SpO 2 or PaO 2 values disproportionate to dyspnea and/or radiological evidence of lung injury on chest radiograph or chest CT, has been widely reported but has never been observed in classic non-COVID ARDS. Various hypotheses have been put forward to explain the silent hypoxemia of COVID-19 lung injury: (1) vascular damage resulting from endotheliitis, with macrothrombi and microthrombi more pronounced in COVID-19 pneumonia compared with other viral or bacterial pneumonias resulting in lung injury , ; (2) diffuse alveolar damage might not be the predominant pathological condition of COVID-19 lung injury leading to hypoxemia because lung compliance compromise is not significant ; (3) pulmonary vascular dilatation and evidence of perfusion in areas of the lung with GGOs on chest radiograph and chest CT as evidence of possible failure of the compensatory mechanism of physiological hypoxic pulmonary vasoconstriction ; and (4) impaired central and peripheral oxygen sensing and dyspnea perception based on the presence of angiotensin-converting enzyme-2 (ACE2) receptors in the carotid body and brain, with potential direct viral effects and accounting for the observation that dyspnea is less prevalent. Other pandemic-causing coronaviruses such as SARS-CoV and MERS-CoV have been shown to infect brainstem respiratory centers leading to respiratory failure. ,
Swenson et al., in their review of silent hypoxemia and its pathophysiology in COVID-19, discussed these four hypotheses and argued that the evidence is not compelling enough to conclude that the pathophysiology of COVID-19 is unique to advocate a different therapeutic approach from the current protocols of care for classic ARDS. The perceived differences and uncertainties in the pathophysiology of COVID-19 lung injury in comparison with the pathological manifestations of classic ARDS are summarized in Table 5.10 , which is adapted from reference 72.
| Pathophysiological Measures | COVID-19 Lung Injury | Non–COVID-19 ARDS | Comment |
|---|---|---|---|
| Vascular regulation | No direct evidence of HPV impairment | Intact vascular responsiveness | Vascular endotheliitis and microthrombi much more prevalent in COVID-19 lung injury |
| Lung compliance | Compliance reduced | Compliance greatly reduced | Clinically nonsignificant difference in reported values of C ST ranges |
| Neural oxygen sensing and dyspnea perception | Impaired central and peripheral oxygen sensing and dyspnea perception postulated based on the presence of ACE2 receptors in carotid body and brain and potential direct viral effects Dyspnea less prevalent | Preserved oxygen sensing at both peripheral and central chemoreceptors with intact dyspnea perception Dyspnea more prevalent | No direct HVR testing has been performed in both groups |
The principal causes of gas exchange abnormalities leading to hypoxemia in COVID-19 are the same as those of other infectious viral and bacterial pneumonias and ARDS: V/Q mismatch and intrapulmonary shunt. As mentioned earlier, endothelial damage, macrothrombi, and microthrombi could be more preponderant in COVID-19–induced hypoxemia. ,
Phenotypes of COVID-19–Associated ARDS
In the early months of the pandemic, Gattinoni et al. described two distinct phenotypes of COVID-19 pneumonia, type L (standing for low elastance, i.e., high compliance), and type H standing for high elastance, that is, low compliance. Type L pneumonia is characterized by the following features: (1) low elastance (preserved compliance) and hence normal aeration/ventilation; (2) low V/Q ratio and hypoxia, possibly resulting from impairment of hypoxic pulmonary vasoconstriction; (3) low lung weight because GGOs are found predominantly in the subpleural regions; and (4) low lung recruitability because most alveoli are ventilated. Type H pneumonia showcases classic ARDS features: (1) high elastance (greatly reduced compliance) and severely compromised ventilation, (2) high right-to-left shunt resulting from perfusion of nonventilated regions, (3) high lung weight resulting from alveolar edema; and (4) high recruitability resulting from nonventilated regions. The phenotype categorization has implications for respiratory management; however, type L could be an earlier manifestation in the timeline of the evolution of COVID-19 lung injury.
COVID-19 Lung Injury Versus High-Altitude Pulmonary Edema
There have been some comparisons of lung involvement in COVID-19 with HAPE, so it is appropriate to look into the similarities and differences between the two pathological conditions because it has implications for clinical management of COVID-19 lung injury. HAPE and COVID-19 pneumonia are both not of cardiogenic origin, which is because of left heart decompensation with elevation of left atrial pressure, for example, from mitral/aortic valve stenosis. Both HAPE and COVID-19 lung injury are characterized by evidence of fluid accumulation in the interstitial space and alveoli, visualized as bilateral opacities on chest radiographs and chest CT. However, the similarities end there because the basic pathophysiological mechanisms of pulmonary edema are different for the two conditions. The differences in the pathophysiology of HAPE and COVID-19 lung injury are summarized in Table 5.11 . HAPE can be typically managed by supplemental oxygen or descending to lower elevations. This intervention quickly increases alveolar partial pressure of alveolar oxygen (PO 2 ) and PaO 2 (partial pressure of arterial oxygen) leading to a reversal of the basic pathophysiology of HAPE: (1) reduction in hypoxic pulmonary vasoconstriction, (2) lowering of pulmonary arterial pressure, and (3) a reduction in hydrostatic pressure responsible for interstitial and alveolar fluid accumulation. However, an increase in FiO 2 alleviates the hypoxemia of COVID-19 but cannot reverse the pathological changes of diffuse alveolar damage, endotheliitis, and thromboembolic changes of COVID-19 lung injury.
| Pathophysiological Measures | High-Altitude Pulmonary Edema | COVID-19 Lung Injury |
|---|---|---|
| HPV and pulmonary hypertension | High pulmonary pressure (40–60 mm Hg) HPV occurs unevenly Increase in capillary hydrostatic pressure leads to interstitial and alveolar edema (overperfusion edema) | HPV absent Mild pulmonary hypertension (25–30 mm Hg) |
| Alveolar fluid clearance | Sodium transport decreased Reduction in alveolar fluid reabsorption | Alveolar epithelial fluid reabsorption impaired |
| Inflammatory changes | No inflammation in early stages No neutrophil infiltration or proinflammatory cytokines Mild alveolar hemorrhage with escape of plasma proteins More of a hemodynamic problem than inflammation | Cytokine mediated injury Neutrophil and macrophage infiltration Pneumocyte death |
| Surfactant | No impairment in production or function | Impaired surfactant production and function |
| Ventilation-perfusion (V/Q) mismatch | Moderate | Severe V/Q mismatch and right to left shunt |
| Thromboembolic changes | Not observed | Microthrombi and embolism |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


