Puberty and its disorders in the male
PRENATAL NEUROBIOLOGY OF PUBERTY
TESTICULAR DIFFERENTIATION AND DEVELOPMENT
REGULATION OF THE TIMING OF PUBERTY
Genetic Disorders Causing GnRH Deficiency
Genetic Variation in Normal Puberty
External Factors and Secular Trends in the Timing of Puberty
Effect of BMI on Pubertal Timing
GnRH-Dependent Forms of Precocious Puberty
GnRH-Independent Forms of Precocious Puberty
Evaluation of the Boy with Precocious Development of Secondary Sexual Characteristics
Treatment of the Child with Precocious Puberty
OTHER DISORDERS OF THE MALE REPRODUCTIVE ENDOCRINE AXIS
Androgen Receptor (AR) Mutations
Persistent Mullerian Duct Syndrome
Testicular Regression Syndrome (Anorchia), Cryptorchidism, and Hypospadias
EVALUATION OF THE CHILD WITH DELAYED PUBERTY
TESTOSTERONE: THE HYPOGONADAL MALE ATHLETE AND THE INDIVIDUAL WITH A DSD
Puberty is a period during which children attain adult secondary sexual characteristics and reproductive capability.1 In humans, two distinct processes of sexual maturation are recognized: gonadarche and adrenarche. Gonadarche is defined as the growth and maturation of the gonads associated with increased sex steroid secretion. Gonadarche requires an intact hypothalamic-pituitary-gonadal (HPG) axis, and any disruption of this axis can result in temporary or permanent disorders of reproductive endocrine function. Adrenarche is defined as maturation of the adrenal cortex associated with increased secretion of dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEAS), and androstenedione. Unlike gonadarche, adrenarche is a phenomenon limited to humans and some great apes. The physiologic foundations for puberty begin in utero with the development of the neurobiologic structures that govern the hypothalamic-pituitary component of the HPG axis and with the differentiation and the development of the gonads. The entire process, extending from fetal life through achievement of reproductive competence, represents the dynamic and coordinated interactions of an expanding list of genes, proteins, signaling molecules, paracrine factors, and epigenetic events.
Prenatal neurobiology of puberty
In postnatal life, the gonadotropin-releasing hormone (GnRH) neurons are located in the hypothalamus and constitute the GnRH pulse generator, which produces an intermittent discharge of GnRH into the hypophysial portal circulation to stimulate gonadotropin synthesis and secretion by the pituitary gonadotropes. The developmental progression of these neurons is governed by multiple regulatory factors; mutations in some of these factors have been associated with disorders of puberty.2
However, the 1000 to 2000 GnRH neurons originally differentiate in the olfactory placode and begin their migration along the vomeronasal nerves to the cribriform plate around the sixth week of gestation. This migration can be categorized into four stages. The first stage involves the differentiation of the GnRH neurons from a heterogenous stem cell population in the embryonic olfactory placode. Factors involved in these early stages include the proteins encoded by fibroblast growth factor 8 (FGF8), fibroblast growth factor receptor 1 (FGFR1), heparin sulfate 6-O-sulfotransferase 1 (HS6ST1), chromodomain helicase DNA-binding protein 7 (CHD7), and nasal embryonic luteinizing hormone (LH)–releasing hormone (NELF). The extracellular domain of FGFR1, a tyrosine kinase receptor, interacts with heparan sulfate proteoaminoglycan, which functions as its coreceptor. Nonrandom modifications of the sugar moieties associated with heparan sulfate proteoaminoglycans such as by HS6ST1 facilitate FGFR1 function. Binding of its cognate ligand, FGF8, to FGFR1, initiates signaling. The FGF8-FGFR1 signal transduction pathway plays an essential role in the neurobiology of GnRH neurons. Several additional genes show spatiotemporal expression patterns similar to FGF8 and appear to modulate FGF8 signaling through FGFR1. These genes include fibroblast growth factor 17 (FGF17), interleukin 17 receptor D (IL17RD), dual specificity phosphatase 6 (DUSP6), sprout homologue 4 (Drosophila) (SPRY4), and fibronectin leucine rich transmembrane protein 3 (FLRT3).3
Eventually, these neurons reach the hypothalamus where they extend projections to the median eminence to form a network, which completes the third stage of GnRH neuron migration. The last stage comprises functional activity. By the 15th week of human gestation, the GnRH pulse generator is modulating the function of the fetal gonadotropes. The HPG axis is functionally active for the first time during fetal development, and it continues to function in infancy until it enters a relative quiescent state, often referred to as the juvenile pause or prepuberty. Likely sharing some similarities but also having some differences,4 the molecular mechanisms responsible for the prepubertal inactivation of the HPG axis and its reactivation at the onset of puberty remain to be characterized.
Testicular differentiation and development
Prenatal testicular development
A brief summary of prenatal testicular differentiation and development follows; a more detailed description of testicular differentiation and sex development can be found in Chapter 4. Beginning at approximately 4 to 6 weeks of gestation, the primordial bipotential gonad arises from a condensation of the mesoderm of the urogenital ridge. Genes involved in the development of this bipotential gonad include the Wilms tumor (WT1) gene, GATA4, chromobox homologue 2 (CBX2), and steroidogenic factor-1 (NR5A1). During this time, the primordial germ cells proliferate and migrate from the hindgut to colonize the developing gonad. The bipotential gonad is composed of supporting cells, endothelial cells, steroid-secreting cells, and germ cells.
In the usual circumstance, the presence of a Y chromosome bearing the sex-determining region on the Y (SRY) gene promotes testicular differentiation. However, novel data emphasize the complexity of gonadal differentiation with involvement of signaling molecules—for example, SRY-box 9 (SOX9) and Forkhead transcription factor 2 (FOXL2)—that activate or repress gonadal determination factors involved in testicular and ovarian development. Sertoli cell differentiation is the earliest manifestation of testicular differentiation. At approximately 7 to 9 weeks of gestation in the human XY gonad, the Sertoli cells envelope the germ cells to form the seminiferous cords. Curiously, germ cells are not required for initial phases of testicular differentiation. Rather, the local environment directs the developmental fate of the primordial germ cells. During the migration though the hindgut, the primordial germ cells proliferate. The stem cell factor (Steel factor) and c-KIT receptor guide the primordial germ cells to the developing genital ridges. This signaling system ensures that viable primordial germ cells travel to their proper environmental niche and that misdirected primordial germ cells undergo apoptosis. Primordial germ cells that escape apoptosis and migrate to other physical locations, such as the mediastinum or central nervous system, can develop into extragonadal germ cell tumors. Primordial germ cells that occupy the genital ridge become pluripotent gonocytes that express specific stem cell markers including placental/germ cell alkaline phosphatase (PLAP) and octamer binding transcription factor 3/4 (OCT3/4).
Internal genital structures are also bipotential. Sertoli cells secrete anti-Mullerian hormone (AMH), which induces regression of the Mullerian ducts through its actions on the type II AMH receptor. The fetal Leydig cells, initially stimulated by placental hCG, secrete testosterone which stabilizes the Wolffian ducts. In the human male fetus, the testicular compartments, tubular and interstitial components, and specific cell types, Leydig, Sertoli, and germ cells can be visualized by 11 weeks of gestation. The most rapid growth in Sertoli cell number appears to occur during the latter half of the first trimester and the second trimester.5
During early gestation, placental hCG governs Leydig cell proliferation and testosterone and insulin-like factor 3 (INSL3) secretion until endogenous LH regulates these activities in midgestation. Due to this role of placental hCG during early gestation, gonadotropin deficiency does not influence male sexual differentiation. LH secretion influences the number of fetal Leydig cells because the number is decreased in anencephalic fetuses and is increased in 46,XY fetuses, with elevated gonadotropin concentrations secondary to complete androgen insensitivity.6 Follicle-stimulating hormone (FSH) secretion influences Sertoli cell differentiation and AMH and inhibin B secretion.7
Testicular descent occurs in two phases. The transabdominal phase begins at approximately 12 weeks of gestation and is influenced by the Leydig cell product, insulin-like factor 3 (INSL3), and its cognate receptor, leucine-rich repeat-containing G protein–coupled receptor 8 (LGR8). The second androgen-dependent phase, descent of the testes through the inguinal canal, is usually accomplished by the end of week 35.8
Postnatal testicular development
Following a brief perinatal decline, the hypothalamic-pituitary-testicular axis is active during the first few months of life with testosterone concentrations peaking at 1 to 2 months of age.9 By approximately 6 months of age, testosterone concentrations decline to prepubertal levels. Despite the increased neonatal HPG activity, sexual hair does not develop and gametogenesis does not occur due to limited androgen receptor (AR) signaling in certain tissues (e.g., Sertoli cells). Throughout infancy, AR is expressed in Leydig cells, but not in Sertoli cells.10 During infancy and childhood, seminiferous cords are solid and generally filled with immature Sertoli cells. The germ cells are limited to spermatogonia and Leydig cells are rarely visualized.11 After early infancy, which provides a window of opportunity to assess testicular function, hCG stimulation may be needed to assess Leydig cell function. Inhibin B and AMH continue to be secreted during childhood providing valuable markers of Sertoli cell function.
Reactivation of the GnRH pulse generator at the time of puberty stimulates pituitary LH and FSH secretion (Figure 17-1). The LH stimulation leads to increased testicular testosterone and INSL3 secretion. The increased testosterone acts as a paracrine factor to induce Sertoli cell maturation. Pubertal maturation of the seminiferous tubules is characterized by cytoskeletal rearrangements including development of tight junctions, Sertoli cell polarization, Sertoli cell proliferation, migration of spermatogonia toward the basement membrane, and decreased AMH secretion.12 FSH stimulates Sertoli cells to secrete inhibin B, which from midpuberty onward serves as the major negative regulator of pituitary FSH secretion. Prior to puberty, inhibin B is germ cell independent. After puberty, inhibin B secretion becomes germ cell dependent and provides a marker of germ cell depletion in adults.13
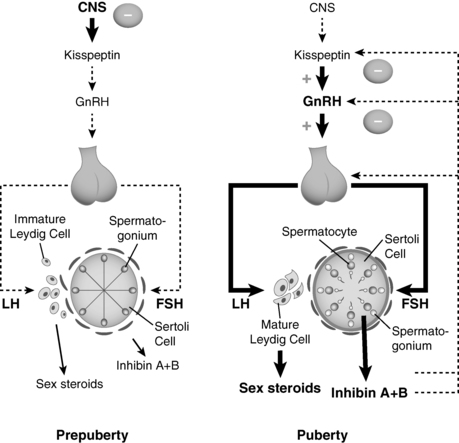
FIGURE 17-1  Testicular products, such as inhibins, play a small role in regulation of the HPG axis prior to puberty, but most of the dampening of the HPG axis after infancy and until the onset of puberty (left panel) derives from central inhibition of the HPG axis. The basis for that inhibition is not fully understood nor is the basis for its diminution, which leads to emergence of central activation (right panel), increased gonadotropin releasing hormone (GnRH) secretion, and the onset of puberty. As pubertal development progresses, lumen develops in Sertoli cells. Inhibin B secreted from the mature testes (right panel) has a more substantial role in regulating/inhibiting hypothalamic-pituitary activity. Weight of lines (from dotted to thin solid to thick solid) indicates increasing strength of the particular signal. LH, luteinizing hormone; FSH, follicle-stimulating hormone.
Testicular products, such as inhibins, play a small role in regulation of the HPG axis prior to puberty, but most of the dampening of the HPG axis after infancy and until the onset of puberty (left panel) derives from central inhibition of the HPG axis. The basis for that inhibition is not fully understood nor is the basis for its diminution, which leads to emergence of central activation (right panel), increased gonadotropin releasing hormone (GnRH) secretion, and the onset of puberty. As pubertal development progresses, lumen develops in Sertoli cells. Inhibin B secreted from the mature testes (right panel) has a more substantial role in regulating/inhibiting hypothalamic-pituitary activity. Weight of lines (from dotted to thin solid to thick solid) indicates increasing strength of the particular signal. LH, luteinizing hormone; FSH, follicle-stimulating hormone.
The increase in Sertoli cell number contributes to the increase in testicular volume that marks the onset of gonadarche in boys. As puberty progresses, the seminiferous tubules are larger and develop a lumen; typical Leydig cells become apparent. Although much variation in chronologic age and testicular volume has been described, spermarche precedes peak pubertal linear growth velocity and generally occurs at a median testicular volume of 10 to 12 mL.14 Beginning in early puberty, testosterone and AMH show an inverse relationship reflecting the paracrine actions of testosterone to repress AMH secretion by Sertoli cells. Intratesticular testosterone and AR expression in Sertoli cells are essential for the decline in AMH concentrations, meiosis, and spermatogenesis.11 Hence, AMH concentrations provide an indication of Sertoli cell function and androgen action in the testes.
The androgen receptor
The androgen receptor (AR) binds androgens to initiate a signal transduction cascade mediating the effects of testosterone and DHT. The AR gene is located at Xq11-q12. Similar to other members of the steroid-thyroid nuclear hormone receptor family, AR (also known as NR3C4) has a modular structure with N-terminal regulatory (NTD), DNA binding (DBD), and ligand binding (LBD) domains. The DBD contains the cysteine residues that coordinate zinc atoms to form the zinc finger domains, which bind to DNA. The LBD is composed of 12 α helixes associated with antiparallel β sheets, which form a tripartite sandwich structure.
Physiology of puberty
Endocrinology
The onset of puberty is heralded by an increase in the secretion of gonadotropin releasing hormone (GnRH) (see Figure 17-1) from a diffusely distributed network of hypothalamic neurons. Intermittent secretion of GnRH into the hypophysial portal circulation stimulates gonadotropes in the anterior pituitary to synthesize and secrete luteinizing hormone (LH) and follicle stimulating hormone (FSH) that, in turn, bind to ligand specific receptors in the Leydig and Sertoli cells of testes, respectively. Gonadotropin stimulation initiates gonadarche, and the production of sex steroids, most notably testosterone, and at later pubertal stages estradiol as well.15 Testosterone and estradiol, together with inhibin, activin, and follistatin, provide signals that regulate the subsequent activity of the hypothalamus and pituitary gland. The transition from prepubertal quiescence to the adolescent pattern of GnRH secretion is a gradual rather than abrupt process. LH and FSH pulsatility has been detected in normal children as young as 4 years of age.16–19 Throughout childhood, GnRH secretion appears to undergo small but progressive increases until the onset of puberty, when GnRH secretion increases, first at night and eventually throughout the day.20,21
Neuroendocrine regulation of pubertal onset
Understanding what factors contribute to the dampening of the HPG axis after infancy and what factors lead to the reemergence of GnRH secretion is critical to understanding the regulation of pubertal timing. Research into kisspeptin (Kiss1) and its receptor (Kiss1R, formerly G protein–coupled receptor 54, GPR54) in both animal and human studies has identified them as critical components of the HPG axis. The first indications of the importance of this signaling complex as a regulator of the HPG axis came in 2003 when two independent groups reported deletions and inactivating mutations of GPR54 in patients with hypogonadotropic hypogonadism (HH).22,23 Subsequently, activating mutations in this pathway were associated with central precocious puberty in the case of a female with an autosomal dominant mutation in GPR54.24 Thus, it is clear that activation of Kiss1R by kisspeptins plays a pivotal role in the onset of puberty.
It is not yet known, however, whether this system is the initial trigger of puberty or whether it acts either as a downstream effector or in concert with other regulatory factors.25,26 For example, the discovery that mutations in TAC3 (encoding neurokinin B) or its receptor TACR3 (encoding NK3R)27 can cause HH has focused much attention on how hypothalamic neurons that coexpress kisspeptin, neurokinin B, and dynorphin (abbreviated as KNDy neurons) regulate the HPG axis in males and females.28–30 Another excitatory factor in the hypothalamus is glutamate, an important stimulator of GnRH secretion through its actions at n-methyl, D-aspartate (NMDA) and kainate receptors. GnRH secretion is stimulated also by factors such as norepinephrine, dopamine, TGFα, neuregulin signaling via erbB4 receptors, leptin, and galanin-like peptide.31–35 Many of these factors likely act via a complex, intricate communication network that exists between glial cells and neurons within the hypothalamus.36 The potential roles that these and other compounds play in regulating the onset of puberty remains an area of active investigation.
Gamma-amino butyric acid (GABA) neurons appear to play a prominent role in inhibiting prepubertal GnRH release.37 There is evidence that GABA can also stimulate GnRH secretion, but this variable action may be dependent on developmental stage, composition of GABA receptors, and expression of KCC2 (a protein that can alter the inhibitory and excitatory properties of chloride channels).38 The finding that neuropeptide Y (NPY) mRNA expression in the hypothalamus of juvenile monkeys is higher than neonatal animals suggests that NPY may play a role in the juvenile pause.4 Other factors that likely inhibit GnRH release include endogenous opioids (i.e., β-endorphin) and melatonin, but neither of these compounds likely plays a major role in regulating the timing of puberty.35
Somatic changes
In boys, the first marker for the onset of puberty is most often the change from Tanner genital stage G1 to stage G2, including enlargement of the testes (i.e., achievement of volume greater than 3 mL or testicular length greater than or equal to 25 mm). Originally Marshall and Tanner reported the mean (SD) onset of puberty in boys to be 11.64 (1.07) years.39,40 These pubertal stages (Figure 17-2) were based on photographic observation of genital development of a longitudinal but still relatively small sample of 228 boys living in a children’s home. Despite the probably poor representative nature of this sample, comparable studies in Switzerland,41 the United States,42 and Denmark43 reported roughly similar mean ages of puberty onset. Although the mean age of onset may be fairly uniform, the onset of puberty occurs across a wide range of ages in normal, healthy adolescents. Several pathologic states influence the timing of puberty either directly or indirectly and contribute to this splay, but the great majority of the variation in pubertal timing cannot be attributed to any clinical disorder. Ninety-five percent of boys experience the onset of genital development between 9.5 and 13.5 years,44,45 data that have led to the traditional definition of sexual precocity in boys as development of secondary sexual characteristics before age 9 years and delayed puberty as lack of testicular enlargement by age 14 years.
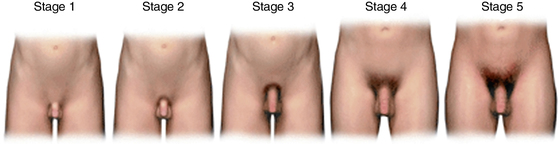
FIGURE 17-2  In boys, genital development is rated from 1 (preadolescent) to 5 (adult); stage 2 marks the onset of pubertal development and is characterized by an enlargement of the scrotum and testis and by a change in the texture and a reddening of the scrotal skin. Pubic hair stages are rated from 1 (preadolescent, no pubic hair) to 5 (adult), and stage 2 marks the onset of pubic hair development. Although pubic hair and genital development are represented as synchronous in the illustration, they do not necessarily track together and should be scored separately. (Reproduced with permission from Carel, J. C., & Leger, J. [2008]. Clinical practice: precocious puberty. N Engl J Med, 358, 2366–2377.)
In boys, genital development is rated from 1 (preadolescent) to 5 (adult); stage 2 marks the onset of pubertal development and is characterized by an enlargement of the scrotum and testis and by a change in the texture and a reddening of the scrotal skin. Pubic hair stages are rated from 1 (preadolescent, no pubic hair) to 5 (adult), and stage 2 marks the onset of pubic hair development. Although pubic hair and genital development are represented as synchronous in the illustration, they do not necessarily track together and should be scored separately. (Reproduced with permission from Carel, J. C., & Leger, J. [2008]. Clinical practice: precocious puberty. N Engl J Med, 358, 2366–2377.)
Development of secondary sexual characteristics results from both gonadarche and adrenarche. Adrenarche refers to the maturation of the zona reticularis of the adrenal gland, resulting in increased production of adrenal androgens associated with secondary sexual characteristics, such as the development of pubic hair (pubarche), axillary hair, body odor, and acne. Like gonadarche, the onset of adrenarche appears to be a gradual, progressive maturational process that begins in early childhood and is marked by the further increases of production of adrenal androgens (DHEA, DHEA-S, and androstenedione) around the time of puberty.46 Adrenarche sometimes precedes gonadarche by 1 to 2 years in boys and girls, but the timing of clinical signs can vary. Although adrenarche and gonadarche often overlap, they are separate processes that are independently regulated.47,48 The triggers for adrenarche remain unknown; however, alterations in body weight and body mass index as well as in utero and neonatal physiology likely modulate this developmental process, perhaps along with intra-adrenal cortisol production.49–51
Regulation of the timing of puberty
Genetics
The timing of puberty is influenced by both genetic and environmental factors.52,53 Evidence for the genetic contribution is provided by correlations in timing within families and between monozygotic twins. These data derive mostly from studies of females and suggest that more than half of the variation in the most commonly used marker for the timing of puberty, age at menarche, is attributable to additive genetic effects and the rest appears to be attributable to nonshared environmental effects.34,52,54–61 It is important to note that this genetic component does not preclude a significant role for environmental influences that may have changed over time. However, despite changing environmental and secular influences, genetic background still plays a significant role in regulating the variation of pubertal timing within a population at any particular point in time.
Genetic disorders causing GnRH deficiency
The study of genetic disorders that cause GnRH deficiency, such as isolated hypogonadotropic hypogonadism (HH) and Kallmann syndrome (KS), has significantly increased our understanding of development and function of the hypothalamic pituitary gonadal (HPG) axis.59,62–67 These studies have led to the identification of many genes that play critical roles in the HPG axis. For example, this work has defined roles for the genes that lead to HH (GNRHR, GNRH1, GPR54, NELF, FGFR1, FGF8, PROK2, PROKR2, TAC3, TACR3, CHD7, and HS6ST1), to X-linked (KAL1) and autosomal (NELF, FGFR1, PROK2, PROKR2, FGF8, WDR11 and CHD7) forms of KS, to obesity and HH (LEP, LEPR, and PC1), and to abnormal HPG development (DAX1, SF-1, HESX-1, LHX3, and PROP-1). Moreover, research in this field permits recognition that, in some cases, HH and KS can be caused by mutations in the same genes and derive from a monogenic or oligogenic basis.68 In contrast to this great progress, the specific genetic factors that regulate the variation in pubertal timing in the general population are just emerging. In addition to genes identified through genome wide association studies (discussed later), another factor may be the makorin RING-finger protein 3 (MKRN3). Loss-of-function mutations in MKRN3 were associated with GnRH-dependent precocious puberty.69
Normosmic hypogonadotropic hypogonadism
Although several HH-related genes have been identified, HH with normal olfaction has been primarily associated with mutations in the genes for the gonadotropin-releasing hormone receptor (GNRHR) and the KISS1 (kisspeptin) receptor (KISS1R, formerly GPR54).22,23,70–74 Estimates regarding the frequency of GNRHR mutations in normosmic HH range from 3.5% to 10.4%.75,76 Mutations in GNRH1 have also been identified in patients with normosmic HH but only in rare case reports.77,78 Although these individuals have normal olfaction, they can have other nonreproductive clinical features such as midline facial defects, renal agenesis, and skeletal abnormalities that may provide clues about the underlying pathophysiology.67 Interestingly, a case of constitutional delay of growth and puberty (CDGP) has also been associated with a homozygous partial loss-of-function mutation in GNRHR,79 and pedigrees of probands with HH can include individuals with delayed but otherwise normal puberty. However, more extensive analyses suggest that genetic variation in neither GNRH1 nor GNRHR nor other HH-related genes is a common cause of late puberty in the general population80,81; whether combinations of rare variants in KS or HH-related genes is a cause of some cases of constitutional delay of growth and puberty remains to be determined.
Mutations in genes encoding neurokinin B and its receptor, TAC3 and TACR3, have been identified in HH patients.27 As noted earlier, these genes are highly expressed in the same neurons that express kisspeptin, further emphasizing the role of kisspeptin in the regulation of pubertal timing.
Kallmann syndrome
HH associated with abnormal sense of smell (anosmia/hyposmia) is referred to as Kallmann syndrome. As with cases of HH and normal olfaction, other nonreproductive clinical features can be seen in individuals with KS, including ichthyosis, choanal atresia, horseshoe kidneys, and mirror movements of the hands (synkinesia).67 Several genes critical to HPG axis function and olfactory development have been identified through investigation of Kallmann syndrome. In particular, mutations in Kallmann syndrome 1 (KAL-1)82,83 and fibroblast growth factor receptor 1 (FGFR1)84 have been implicated in the X-linked and autosomal dominant forms of the disease, respectively, and appear to account for approximately 20% of patients with KS.85 Mutations in the prokineticin receptor-2 gene (PROKR2), a G protein–coupled receptor, and in its ligand prokineticin-2 (PROK2) have also been identified in patients with KS,85 demonstrating that prokineticin signaling is important for olfactory and HPG axis development. One of the patients in this initial series was heterozygous for both a PROKR2 mutation and a KAL1 mutation, suggesting a possible digenic mode of inheritance.85 Finally, mutations in the nasal embryonic luteinizing hormone releasing hormone factor (NELF), which plays a role in migration of GnRH neurons and olfactory axon outgrowth,86 have also been implicated in the pathogenesis of KS.87 A heterozygous deletion in NELF was initially reported as a component, along with FGFR1, of digenic KS,88 but it has recently been reported that NELF can lead to HH or KS via monogenic as well as digenic inheritance.89
The distinction among the different abnormalities of pubertal development is not always clear cut. For example, a comprehensive study of PROK2 and PROKR2 in HH and KS patients found mutations in both genes distributed in both groups of patients.90 Mutations in the FGF8 gene, which encodes a ligand for FGFR1, have been observed in HH patients accompanied by variable olfactory phenotypes.91 Mutations in CHD7, a gene responsible for CHARGE syndrome, which shares some developmental features with KS, have been identified in patients with both normosmic HH and KS.92,93 More recently, mutations in FGFR1, FGF8, and PROKR2 have been identified among subjects with combined pituitary hormone deficiency (CPHD) and septo-optic dysplasia (SOD), indicating that in some cases HH and CPHD/SOD can share genetic etiologies.94,95 The distinction between HH and constitutional delay of growth and puberty (CDGP) is also not always clear. Variation in FGFR1 does not appear to be a major cause of CDGP,81 although loss-of-function mutations in FGFR1 can cause delayed puberty in members of HH pedigrees.96,97 Instances of reversible HH have also been reported,98 raising the possibility that some cases of HH may represent severe versions of CDGP, further blurring the distinction between HH and CDGP.
Other genes associated with HH
Leptin appears to act as a permissive factor in pubertal maturation.34 HH (accompanied by obesity) can result from defects in the leptin (LEP) or the leptin receptor (LEPR) genes, highlighting the importance of nutrition in modulating the HPG axis. The neuronal targets for leptin’s action are incompletely explained because leptin receptors are not expressed by GnRH neurons, suggesting that the site of leptin action may be upstream of the GnRH neurons. However, association studies have found no substantial association between common polymorphisms in LEP and LEPR and constitutional delay of growth and puberty (CDGP) or age at menarche in the general population.81,99
Mutations in several pituitary transcription factors, including HESX-1, LHX3, and PROP1, can lead to combinedpituitary hormone deficiencies that include HH as a phenotype. The gene for prohormone convertase-1 (PC-1) has been associated with obesity and HH, possibly as a result of defective processing of neuropeptides or prohormones that are components of GnRH secretion.66,100 Other causes of HH include mutations in genes that are critical to HPG development. This category includes the genes for the orphan nuclear receptors DAX1 (dosage-sensitive sex reversal adrenal hypoplasia critical [DSS-AHC] region on the X chromosome, gene 1) and steroidogenic factor-1 (NR5A1).
Genetic variation in normal puberty
Observations regarding the greater concordance in age at pubertal development in monozygotic twins and correlation at age of menarche in mothers and daughters emphasized the role of genetic influences in the timing of normal pubertal development.101 Approaches utilized to identify specific genetic factors include candidate genes studies and genome-wide association (GWA) studies.
Candidate gene-based studies
In an association study that tested for associations between common variants in 10 HH-related genes (GNRH1, GNRHR, KISS1R/GPR54, KISS1, LEP, LEPR, FGFR1, KAL1, PROK2, and PROKR2) and age at menarche, only nominally significant associations between SNPs in several of the genes and age at menarche were identified, indicating that genetic variation in these 10 genes does not appear to be a substantial modulator of pubertal timing in the general population.81
Other work has also shown no evidence for substantial association between SNPs in GNRH1 and GNRHR80 or LEP and LEPR99 and alterations in pubertal timing. In a direct sequencing study (which assessed variation in FGFR1, GNRHR, TAC3, and TACR3 in 146 Finnish subjects), variants in the coding regions of these genes were not identified as likely causes of constitutional delay of growth and puberty in the general population.102
Genome-wide association (GWA) studies
Common variants in LIN28B were associated with age at menarche in four independent GWA studies and one meta-analysis.103–107 LIN28B is a human homologue of lin-28, which in C. elegans controls the rate of progression from larval stages to adult cuticle formation, indicating conservation of specific micro-RNA regulatory mechanisms involved in developmental timing.104 These GWA studies involved between 17,000 and 25,000 individuals, all of European descent. In each case, age at menarche (AAM) was analyzed, but in one study107 additional phenotypes (breast development in girls, voice breaking and pubic hair development in boys, and tempo of height growth in both boys and girls) were found to associate with variants in LIN28B, suggesting that control of pubertal timing in boys and girls shares some common elements. One study found that the signal at LIN28B could be split into two haplotypes, suggesting that either multiple variants may associate with AAM at this locus or that a SNP that has not yet been tested for association may represent the true association signal.106 Effect sizes were estimated at approximately 1.2 months earlier per effect allele for LIN28B.105,107 A second menarche locus was identified in two of the four studies at 9q31.2.103,105 The biology behind the locus at 9q31.2 remains unknown, but its effect size is similar to the locus in/near LIN28B.105 The associated SNPs lie in an intergenic region with no obvious candidate genes nearby. The closest gene is TMEM38B, a transmembrane protein gene, which lies approximately 400 kb away from the signal at 9q31.2.105 Although these studies were ground breaking, the LIN28B and 9q31.2 loci together explain only 0.6% of the variance in age at menarche.103
The genetic contribution to the variation in age at menarche was further investigated by a meta-analysis of 32 genome-wide association studies including 87,000 women.104 Thirty new AAM-associated loci were identified, but pubertal timing among males was not assessed. Despite the large size of the meta-analysis, these loci explained only 3.6% to 6.1% of the variance in the age at menarche.104 It is important to note that the small effect size does not negate the importance of the discovery. Findings from GWA studies have highlighted biologic pathways involved in a variety of phenotypes, both through “rediscovery” of genes known to be important and through identification of previously unsuspected pathways.108 This principle also holds true for pubertal timing, with much new biology to explore as the mechanisms through which these newly identified genes modulate pubertal timing are investigated and as future studies determine whether the same genes regulate pubertal timing in boys and in girls.
GWA studies are designed to assess the contribution of common genetic variants to a particular phenotype. However, it is likely that other forms of inheritance also underlie CDGP, including rarer variants (frequency < 5% in the general population) with large or even small phenotypic effects; combinations of variants within a single gene or multiple genes (oligogenicity); structural variation, such as copy number variants; and epigenetics. Indeed, some of these mechanisms have been identified as causes of hypothalamic amenorrhea.109
External factors and secular trends in the timing of puberty
Although these genetic advances are exciting, genetic factors cannot explain the reported secular changes in pubertal timing (discussed later) that have occurred since the late 20th century. Clearly, changes in lifestyle or environmental factors must be involved, likely as independent regulators but also as factors that mediate effects through gene by environment (G X E) interactions. Variables such as increased adiposity, insulin resistance, physical inactivity, psychological factors, and changed dietary habits have all been implicated as possible mediators of the observed change in pubertal timing.52,110,111
The mean age of menarche in mid-19th century Europe was likely between 17 and 18 years.52 Starting from the late 19th century to the mid-20th century, a gradual decline in age at puberty has been reported, more convincingly in girls than in boys,52,112 after which this trend may have slowed. The change in the timing in puberty has likely been the result of better hygiene and nutrition as well as increased stability in socioeconomic conditions.
The extent to which age at puberty has declined in males is controversial. In the mid-1990s, data from the Third National Health and Nutrition Examination Survey (NHANES III), where genital ratings were performed by visual inspection, reported earlier age at puberty in both boys and girls113–116 than what previously had been reported from the United States.44,115,117 Using the traditional cutoff of 9 years, these data suggest that an increased number of boys would be classified as having precocious puberty. However, due to lack of data on pubertal onset in the previous population-based study (Third National Health Examination Survey [NHES III]),44 some controversy remained as how to interpret the NHANES III findings.113,114,116 Furthermore, questions have been raised regarding the criteria used for genital staging in NHANES III.118 A subsequent secular trend analysis between NHES III (which lacked data from the early pubertal stages) and NHANES III did not find clear evidence supporting earlier age at puberty, although some indications were present in non-Hispanic white boys.115 These data were also reviewed by an expert panel, which concluded that the available data are insufficient in quality and quantity to confirm a change in pubertal timing in U.S. boys.112 Conversely, at the same time in Europe, in comparison with NHANES III studies, some data were reported suggesting older ages at pubertal onset in boys.41,119–121 Only a few European studies contained data to assess secular trend in the timing of puberty in Europe, and those do not support a substantial enough change in the age at pubertal onset in boys from the mid-1960s to the late 1990s to warrant a change in the age definitions for precocious and delayed puberty.43,120
Effect of BMI on pubertal timing
Among white American children aged 6 to 11 years, rates of obesity have increased from approximately 5% between 1963 and 1965 to 12% in 1999–2000.122 The possibility that the increasing rates of obesity contributed to the secular trend toward early puberty onset was originally highlighted in 1997.123 However, despite strong evidence in support of a link between increased adiposity and early onset of pubertal markers in girls,124,124 data in boys have remained somewhat ambiguous, with increased body mass index (BMI) being associated in general with earlier puberty but with obesity being associated with delayed puberty in some cases.125,126
Several population studies regarding pubertal timing and BMI have been conducted in boys. A retrospective study of 1520 men with serial height and weight measurements between the ages of 9 and 18 years reported that boys with high childhood BMI tended to have an earlier puberty, whereas boys with a later puberty tended to be taller and were less obese as adults than those who had experienced an earlier puberty.127 Similarly, high BMI gain during childhood has been related to an earlier onset of puberty and reduced height gain during adolescence.128
In a study of 463 Danish choir boys, a significant downward trend in age at voice break was found over a 10-year period (from 14 years to 13.7 years).129 Age at voice break was significantly different between boys in the different prepubertal BMI quartiles, and a trend toward earlier voice break was associated with increasing BMI SDS. Boys in the heaviest quartile at 8 years of age had an increased likelihood of earlier voice break compared with those in the thinnest quartile, suggesting a relationship between prepubertal BMI and the timing of puberty in boys.129
Another Danish study included an evaluation of secular trends in pubertal onset over a 15-year period and their relationship to BMI in boys.121 In a total of 1528 boys, onset of puberty, defined as age at attainment of a testicular volume > 3 mL, occurred 3 months earlier in 2006–2008 than in 1991–1993. Significantly higher levels of luteinizing hormone but not testosterone were found in boys between 11 and 16 years of age in 2006–2008 compared with boys of the same age in 1991–1993. BMI SD scores increased significantly from 1991–1993 to 2006–2008 as well. Interestingly, pubertal onset and levels of luteinizing hormone were no longer significantly different between study periods after adjustment for BMI. In this study, the estimated mean age at onset of puberty had, therefore, declined during the 15-year interval, and this decline was associated at least partly with an increase in BMI.121
In a study from Jamaica, the effects of birth size, growth rates throughout childhood, and body composition on the timing of onset of puberty were assessed in both boys and girls.130 Fast weight gain from age 0 to 6 months and during childhood, but not large birth size, was associated with advanced puberty in both sexes. In addition, elevated fat mass at 8 years of age was associated with advanced puberty in both sexes. These data support the hypothesis that fast growth throughout childhood, especially with fat-mass accretion, is associated with advanced pubertal development.130,131
In a study from Germany, body weight, height, peak height velocity, and pubertal stages were evaluated in 1421 peripubertal children.132 In contrast to the findings from other studies, the researchers found no significant differences in mean pubic hair stage in girls and boys with obesity when compared with either lean or normal-weight children and, when analysis was restricted to children at pubic hair stage 2, age at this stage was found not to differ significantly between normal-weight and obese individuals. In boys, testicular volume at a given age was also similar across all weight groups.133
Effect of endocrine disrupting chemicals
Some evidence supports associations between human pubertal timing and exposure to environmental modifiers, although much of the data relate to girls. Earlier menarche and pubarche have been associated with exposure to polybrominated biphenyls (PBB) and dichlorodiphenyl dichloroethene (DDT), whereas delayed breast and pubic hair development as well as delayed menarche have been associated with lead exposure.134,135 In addition, elevated serum levels of a mycotoxin (zearalenone) have been reported in girls with precocious puberty,136 and phthalate exposure has been associated with changes in pubertal timing. A study in Denmark demonstrated that delayed pubarche, but not thelarche, was associated with high phthalate excretion in urine samples from healthy school girls, which may suggest antiandrogenic actions of phthalates.137 Greater exposure to pesticides has been reported in boys with cryptorchidism or hypospadias.138
The extent to which these effects are associations versus cause-and-effect relationships is unknown as is full understanding of underlying mechanisms. To what extent any effects are shared versus different in girls compared to boys is also not known. Finally, what effects these exposures have in the general population as opposed to isolated examples of associated abnormalities is not clear. Certainly more research is this area is needed.139,140
Precocious puberty
GnRH-dependent forms of precocious puberty
The most common form of precocious puberty is the activation of pulsatile GnRH secretion 2 to 2.5 SDs earlier than average, recognizing that the normal limits for the onset of puberty can vary by geographic area and by ethnicity. This form of precocious puberty is called central or gonadotropin-dependent precocious puberty, and it represents true pubertal development. Central precocious puberty (CPP) may result from hypothalamic tumors or central nervous system (CNS) lesions (neurogenic CPP) but in most cases remains unexplained (idiopathic CPP) (Table 17-1).141,142
TABLE 17-1
Common Etiologies of Sexual Precocity in Boys
| Central (GnRH Dependent) | Peripheral (GnRH Independent) |
| Idiopathic | Congenital adrenal hyperplasia |
| Central nervous system tumors | McCune-Albright syndrome |
| —Hamartomas | Testosterone producing tumors |
| —Astrocytomas | —Adrenal carcinoma or adenoma |
| —Adenomas | —Leydig cell tumor |
| —Gliomas | Gonadotropin/hCG producing tumors |
| —Germinomas | —Choriocarcinoma |
| Central nervous system infection | —Dysgerminoma |
| Head trauma | —Hepatoblastoma |
| Iatrogenic | —Chorioepithelioma |
| —Low-dose CNS radiation | —Teratoma |
| —Chemotherapy | —Gonadoblastoma |
| —Surgical | Exogenous exposure to androgen |
| Malformations of central nervous system | Familial male limited precocious puberty |
| —Arachnoid or suprasellar cysts | Hypothyroidism (Van Wyk–Grumbach syndrome) |
| —Hydrocephalus |
Modified from Nathan BM, Palmert MR (2005). Regulation and disorders of pubertal timing. Endocrinol Metab Clin North Am 34:617-641, ix.
Hypothalamic hamartomas are an example of a cause of neurogenic CPP. The hamartomas are congenital malformations characterized by heterotropic gray matter, neurons, and glial cells, and they are generally located on the floor of the third ventricle or attached to the tuber cinereum. On MR imaging, hamartomas exhibit an isodense fullness. Histologic examination has shown immunoreactivity for GnRH and for astroglial factors such as TGFα. Postulated potential mechanisms include increased GnRH secretion from neurons emancipated from suppression or that TGFα stimulates GnRH secretion from GnRH neurons. Most hypothalamic hamartomas are sporadic, but they may occur in association with Pallister-Hall syndrome, which is due to mutations in the GLI3 gene. Optic gliomas, which can be associated with NF1, can also cause GnRH-dependent precocious puberty.143 Other etiologies of neurogenic CPP include pineal tumors, suprasellar cysts, previous head trauma, CNS radiation, and static encephalopathy.144
Loss-of-function mutations of the makorin RING-finger protein 3 (MKRN3) located at chromosome 15q11-q13 have been associated with GnRH-dependent precocious puberty. All clinically affected individuals inherited the mutated allele from their fathers, which can be explained because this gene is imprinted and expressed only by the paternal allele. Curiously, almost half of these patients were boys; this differs from the typical female predominance of GnRH-dependent precocious puberty. The finding of MKRN3 as a cause of CPP is also interesting because it calls attention to the importance of inhibitory factors in the regulation of pubertal timing (see Figure 17-1) because loss of function of this gene (and loss of a presumed inhibitory role) leads to CPP.69
GnRH-independent forms of precocious puberty
Precocious development of secondary sexual characteristics may also be caused by mechanisms that do not involve activation of pulsatile GnRH secretion. These forms of precocity are called gonadotropin-independent or peripheral precocity and include gonadal and adrenal tumors, tumors producing human chorionic gonadotropin, mutations activating the gonadotropic pathway, and exposure to exogenous sex steroids (see Table 17-1). Familial male-limited precocious puberty (FMPP, OMIM ID: 176410), also known as testotoxicosis, is a rare dominant form of gonadotropin independent precocity caused by constitutively activating mutations of the human LH choriogonadotropin receptor (LHCGR).145 This disorder usually presents by age 1 to 4 years with physical signs of puberty, rapid virilization, growth acceleration, skeletal (bone age) advancement, and elevated testosterone levels despite prepubertal levels of LH.146,147 McCune-Albright syndrome (MAS, OMIM ID: 174800) is another rare cause of male sexual precocity of genetic origin. It is caused by a postzygotic somatic activating mutation of the GNAS1 gene. The mosaic constitutive activation of the GSα protein signaling leads to autonomous cell proliferation and scattered hyperfunction in endocrine organs with a wide phenotypic spectrum.148,149 The classical features include the clinical triad of bone fibrous dysplasia (BFD), café-au-lait skin spots, and precocious development of secondary sexual characteristics. In addition, excessive pituitary function (such as hyperthyroidism due to activation of thyroid-stimulating hormone [TSH] secretion and hypercortisolism due to constitutive activation of ACTH secretion), kidney phosphate wasting, cholestasis, and hypertrophic heart disease can be present.148,150 For reasons that are not clear, MAS leads to sexual precocity more often in girls than in boys.
In 1960, Van Wyk and Grumbach first described a syndrome characterized by breast development, uterine bleeding, and multicystic ovaries in the presence of long-standing primary hypothyroidism.151 A unique diagnostic feature of the Van Wyk–Grumbach syndrome is the combination of delayed bone age with apparent sexual precocity. Boys with this syndrome have macroorchidism without significant virilization. Testicular histology shows enlargement of the seminiferous tubules without an increase in Leydig cell number.152,153 Typically, GnRH stimulation shows a prepubertal response with suppressed LH confirming gonadotropin-releasing hormone (GnRH)-independent precocious puberty. Most cases appear to originate from autoimmune thyroid disease, but there are some case reports where the syndrome is secondary to unrecognized congenital hypothyroidism.154 The pathophysiology of Van Wyk–Grumbach syndrome involves a complex mechanism, which is, at least in part, mediated by the direct action of TSH on FSH receptors. Recombinant human TSH (Rec-hTSH) elicits a dose-dependent cAMP response in cells expressing the human FSH-receptor in vitro; however, the concentration of rec-hTSH required for half-maximal stimulation was several logs greater than that of hFSH.152 Early recognition and initiation of thyroid hormone replacement can lead not only to resolution of symptoms and improvement in adult height but also to avoidance of further diagnostic procedures, fear of malignancy, and unnecessary surgery.
Androgen secreting tumors are rare causes of GnRH-independent precocious puberty in boys. Leydig cell tumors secrete testosterone. Because these tumors are usually unilateral, testicular volume may be asymmetric. The majority of Leydig cell tumors are benign. Ultrasound may be useful because the tumor may be too small to palpate. Mutations in the LHCGR gene have been identified in some adenomas.155 In boys, tumors secreting hCG stimulate testicular testosterone secretion. These tumors are generally hepatic in origin. Virilization is a common presentation for adrenal tumors in children.
The classification of precocious puberty is not always clear. As noted earlier, treatment of peripheral forms of sexual precocity, such as CAH or testicular or adrenal tumors, can lead to true central precocious puberty through subsequent activation of pulsatile GnRH secretion. In addition, in some cases of precocious pubertal development among girls, pubertal manifestations will regress or stop progressing, making treatment unnecessary; such cases are not as commonly seen in boys.156,157 The mechanisms responsible for these nonprogressive forms of precocious puberty are not known. There is evidence that in some cases the HPG axis is intermittently but not fully activated.157,158 Another form of early development of secondary sexual characteristics occurs when the hypothalamic-pituitary-adrenal (HPA) axis matures 1 to 2 years before the HPG axis, causing premature adrenarche. Premature adrenarche, more common among girls, is not associated with progressive pubertal development and is manifested by pubic and axillary hair with modest elevation of DHEA-S, but without substantial advancement in bone age and hence does not require treatment.
In cases of progressive central or peripheral precocity, the concerns include short adult stature due to early epiphyseal fusion and adverse psychosocial outcomes.141,159 Several studies have assessed adult height in subjects with a history of precocious puberty. Mean heights range from 151 to 156 cm in boys and from 150 to 154 cm in girls in older patient series, which corresponds to a loss of more than 20 cm in boys and 12 cm in girls from predicted adult height.160 Height loss due to precocious puberty is inversely correlated with the age at the onset of puberty, and treated patients today tend to have a later onset of puberty than did patients in historical series.160 It is important to assess carefully the growth rate and skeletal maturation of individuals with central precocious puberty due to CNS lesions (e.g., tumors, cerebral malformations or injury from trauma or radiation) because these lesions may be associated with concomitant growth hormone (GH) deficiency, which can be masked by sex steroid–driven adequate growth rates. In such cases, undiagnosed and untreated GH deficiency may result in severely compromised adult height.
Although available data derive mostly from study of girls with early puberty, it has been suggested that a higher proportion of early-maturing adolescents engage in exploratory risky behaviors (sexual intercourse and legal and illegal substance use) at an earlier age than adolescents maturing within the normal age range or later.161,162 However, available data regarding potential adverse psychosocial outcomes specific to patients with precocious puberty are limited, as it is not clear that data obtained from individuals with maturation at the early end of the normal spectrum are fully applicable to precocious puberty.
Pathologic entities leading to sexual precocity warrant treatment, but whether and at what ages one should initiate treatment for idiopathic central precocious puberty is less certain. Currently, robust data regarding the short-term and long-term psychosocial sequelae of CPP and data regarding whether treating CPP with gonadotropin releasing hormone analogues (GnRHa) alter these outcomes are lacking.163 Thus, one needs to be cautious when using psychosocial outcomes as a rationale for pharmacologic intervention to halt pubertal progression, especially among boys with the onset of puberty close to the normal range.
Evaluation of the boy with precocious development of secondary sexual characteristics
Boys with sexual precocity require careful evaluation because many have underlying disorders.141 Those with premature pubarche (early development of pubic hair or body odor) need to be assessed for peripheral causes of precocity before determining that the premature pubarche stems simply from premature adrenarche. Premature adrenarche may be distinguished from true precocity over time by lack of progression. Conversely, progressive precocity is marked by significant bone age advancement (> 2 SD for age), a history of growth acceleration, and a progression of secondary sexual characteristics on physical examination.
The evaluation of a boy with sexual precocity is outlined in Box 17-1. Considerations include first verifying that the pubertal development is occurring outside the range of normal development prior to initiating an evaluation. It is also important to note that all tests are not appropriate in each case and that the diagnostic yield of each test is not known. Hence, it is important to allow history and physical examination to guide the evaluation. The child with bilateral enlarged testicles, for example, is most likely to have central precocious puberty, with FSH having led to the expansion of seminiferous tubule volume (although cases of hypothyroidism can also present with bilateral testicular enlargement). Conversely, the child with bilateral prepubertal sized testicles is more likely to have peripheral precocity, whereas the child with unilateral testicular enlargement may well have a testicular tumor. Testing should be directed accordingly.
Peripheral causes of sexual precocity are characterized by suppressed LH and FSH values in the setting of elevated sex steroid levels. Testosterone levels will be elevated in instances of isosexual precocity (secondary sexual characteristics consistent with male gender), whereas estradiol levels may be elevated in the rare instances of contrasexual precocity (secondary sexual characteristics inconsistent with gender, such as marked breast development as the presenting sign in a boy). Determining the underlying mechanism for progressive peripheral precocity is important because all instances will result from pathologic conditions or exogenous exposures.164 Levels of androgens that are elevated beyond the range expected for the pubertal stage suggest an adrenal or testicular cause of the precocity. DHEA-S is often used as a screen for adrenal tumors or adrenal pathology. Determination of the 17-hydroxyprogesterone concentration is used to screen for congenital adrenal hyperplasia due to 21-OH deficiency. Congenital adrenal hyperplasia and hormone secreting tumors of the adrenal are discussed in detail in Chapter 13.
Historically, the gold standard for the diagnosis of central precocious puberty was GnRH stimulation testing and the demonstration of pubertal gonadotropin responses. However, with the advent of ultrasensitive gonadotropin assays,165 the recognition that these assays can identify individuals with CPP using unstimulated random samples,166 and the unavailability of GnRH for testing has led some to forego stimulation testing. GnRH agonists can be used as an alternative to GnRH for stimulation testing, but again the diagnosis can often be made through the combination of clinical features and a baseline LH value in the pubertal range. Although determining the diagnostic cutoff for the basal LH level is difficult because of a lack of normative data and variability among assays, a value of ≥ 0.3 IU/L using an ultrasensitive assay with a detection limit near 0.1 IU/L is commonly cited.163 If stimulation testing is done, a diagnostic cutoff of 5 IU/L for peak value has been suggested. LH values are more useful than FSH values in the diagnostic evaluation of precocious puberty, but stimulated LH/FSH values can help to identify patients with slowly progressive precocious puberty because these children tend to have FSH predominant responses.157,167 Whether due to referral bias or to differences in underlying physiology, idiopathic central precocious puberty is a more common cause of precocious puberty among girls than among boys in endocrinology clinics.168,169 Hence, all boys with CPP should have brain magnetic resonance imaging (MRI) to exclude underlying pathology.163
In summary, it is important for physicians evaluating patients with suspected precocious puberty to address these questions: Is pubertal development occurring outside the normal range of development? What is the underlying mechanism, and is that mechanism associated with a risk of a serious condition, such as an intracranial lesion? Is the pubertal development likely to progress, and may this impair the child’s normal physical and psychosocial development?
Treatment of the child with precocious puberty
Central precocious puberty
A prerequisite for the consideration of therapy for CPP is the presence of documented, progressive pubertal development over a 3- to 6-month period, although this period of observation may not be necessary if the child is at or beyond Tanner stage 3 genital development. Documentation of progressive development is important because nonprogressive forms of precocious puberty do not require intervention.163 Because epiphyseal fusion is an estrogen-dependent process, early and progressive production of sex steroids can cause rapid advancement of skeletal maturation and result in compromised adult height. Thus, preservation of adult stature is one of the main reasons to consider treatment of CPP with GnRH agonists, which down-regulate the pituitary-gonadal axis and limit pubertal progression. Although the data derive primarily from studies among girls, it appears that the risk of short stature is most pronounced in those children with earlier onset of symptoms and more shortened prepubertal growth periods.170 Other factors, such as advanced bone age, may also contribute to poorer height outcomes.170 In girls, available data suggest that the greatest height gain (preservation) occurs with onset of puberty prior to 6 years of age, with more moderate benefit being seen with onset between 6 and 8 years.163 However, insufficient data exist to draw similar conclusions regarding age of onset and height outcomes among boys171,172; consequently, a consensus conference recommended considering initiation of GnRHa therapy for all boys with onset of CPP before age 9 years who have compromised height potential.163

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


