INTRODUCTION
Over the past decade, insight into the biological basis of prostate cancer development and progression has influenced our approach to treating patients with the disease. While research efforts have historically focused on the prostate cancer epithelial cell, there is growing evidence that interactions between the host tissue microenvironment and the cancer epithelial cell are critical for tumorigenesis. Understanding the bidirectional cancer cell–host interaction now dominates prostate cancer research.
Prostate cancers have recognizable clinical features that allow anticipation of their clinical behavior. Fortunately, the progression from localized, androgen-dependent disease to castration-resistant disease with bone-forming metastases occurs in only a minority of patients. To conceptualize the clinical heterogeneity displayed along this continuum, patients have historically been assigned to different “clinical states” (eg, metastatic castrate vs noncastrate) to help structure treatment recommendations and therapy development (1,2). However, this approach has been limited by the fact that patients within each state display wide biological heterogeneity and response to therapy. To address this, improved classification of the disease based on the underlying molecular mechanisms of progression is necessary. This effort will create a “marker-driven” strategy to more reliably predict prostate cancer progression, permit optimal selection of specific therapies for individual patients, and apply therapy to only those patients who need it. This strategy will favorably improve the outcome of selected patients threatened by their disease while avoiding unnecessary morbidity to the majority who are not.
EPIDEMIOLOGY AND CLINICAL FEATURES
Prostate cancer is a major health-care challenge in the United States (3). It is the second most common cancer in men (behind skin cancer) and the second leading cause of cancer death (behind lung cancer). There are a number of unique clinical features of prostate cancer that distinguish it from other solid tumor types:
Despite the high prevalence of prostate cancer, the majority of patients diagnosed with the disease eventually die from other causes. This is in striking contrast to lung cancer, where the majority of patients diagnosed with the disease die from it.
Cancer of the prostate often has a prolonged natural history. This is evidenced by a high incidence of occult malignancy in autopsy series of men who die from nonprostate cancer causes and in clinically normal prostates of men undergoing cystoprostatectomy for bladder cancer. Therefore, over the course of a normal lifetime, most men will develop “clinically occult” prostate cancer that will never produce symptoms, require treatment, or cause death.
The incidence of detected carcinoma increases with age.
Androgens are a major driving force in normal prostate development and are implicated in tumorigenesis.
Prostate cancer is typically multifocal, commonly presenting as synchronous carcinomas arising in multiple locations, and the malignant potential is determined by the sum of the primary and secondary grades (Gleason score). Thus, biologic heterogeneity is an inherent property of each tumor.
Clinical prostate cancer is more prevalent in Western than Eastern societies, although incidence rates increase for men from China and Japan who immigrate to the United States. This observation implicates environmental factors (diet, lifestyle, etc) in prostate cancer development.
Prostate cancers have a predictable rate and pattern of progression, with bone-forming metastases dominating the clinical progression in the majority of patients with advanced disease. This observation supports the view that the bone-epithelial interaction is central to the progression of prostate cancer.
It has long been recognized that prostate cancer is a disease of the elderly, and epidemiologic data demonstrate that rates of prostate cancer incidence and mortality increase with age (4). While prostate-specific antigen (PSA) screening has led to an earlier average age at diagnosis, mortality is still largely seen in patients 70 years of age or older. As the longevity of populations increases worldwide, the burden of prostate cancer creates a significant health-care challenge. This has generated a sense of urgency among physicians to refine our ability to predict cancer virulence and apply therapy to those patients who need it.
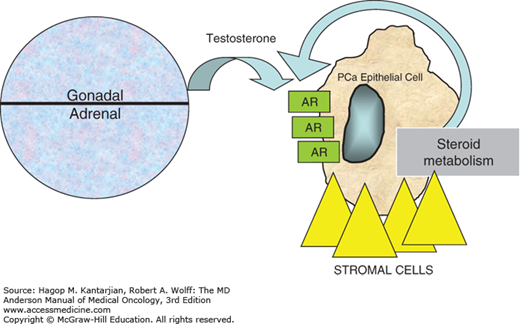
Androgens are central to the normal growth, differentiation, and function of the prostate gland, although the role of androgen receptor (AR) signaling in prostate carcinogenesis and progression has not been fully elucidated. Even in the clinically castrate state (serum testosterone <50 ng/mL), there is growing evidence that prostate cancer cells continue to rely on AR signaling for proliferation (5). One of the principal mechanisms for this involves a gradual shift from endocrine sources of androgens (ie, from the testes and adrenal glands) to paracrine/autocrine/intracrine (ie, intratumoral) sources during prostate cancer progression (Fig. 37-1). This occurs through the peripheral conversion of adrenal steroid precursors (eg, dehydroepiandrosterone and androstenedione) in prostate and bone tissues expressing CYP17 enzymes (6). Other mechanisms accounting for castrate resistance include intratumoral amplification of AR, mutations of AR, changes in levels of AR cofactors, and ligand-independent activation of AR (Fig. 37-1).
Several lines of clinical and experimental evidence support a central role for diet, caloric intake, and obesity in the development of prostate cancer with lethal potential. Obesity, defined as body mass index (BMI) above 30, is manifested by overgrowth of white adipose tissue (WAT). Obesity has been associated with incidence, progression, and mortality of prostate cancer (7,8). The mechanism for the association between obesity and prostate cancer progression is poorly understood. Current models suggest that predetermined genetic traits associated with both obesity and cancers are influenced by lifestyle components such as diet and physical activity. However, epidemiological studies show that cancer can be accelerated in obese patients irrespective of their lifestyle. Thus, it has been proposed that WAT itself may have a direct effect on cancer progression.
Mechanisms linking obesity and aggressive prostate cancer include signaling through insulinlike growth factor 1 (IGF-1) and adipokines (9). The insulin/IGF-1 axis has been widely implicated in obesity-induced tumorigenesis, including prostate cancer (10). Primary human prostate cancer commonly expresses the insulin receptor, suggesting insulin may stimulate tumor growth (11). Both excess body weight and a high plasma concentration of C-peptide are associated with increased prostate cancer–specific mortality (12). In addition, the antidiabetic agent metformin has been shown to reduce all-cause and prostate cancer–specific mortality among diabetic men (13).
Beyond changes in insulin, obesity alters levels of adipokines (such as leptin, adiponectin) due to chronic subclinical inflammation. Leptin is associated with in vitro protumor effect in human prostate cancer cell lines (14), but epidemiologic studies have failed to consistently establish an association with prostate cancer risk and mortality (15). In contrast to leptin, adiponectin serum levels are reduced in obesity and have largely antitumor effects (16).
African Americans have a higher frequency of death from prostate cancer compared to Caucasian and Hispanic Americans. This has variably been attributed to differences in steroid metabolism, genetics, environmental effects, or social factors (17,18). There is a reduced incidence of prostate cancer among Chinese and Japanese Americans, but their incidence is higher than that reported in native Chinese or Japanese persons. Of interest is the fact that northern European men have a higher frequency of prostate cancer than men from southern Europe. A similar finding has been reported in the United States, suggesting that the incidence of prostate cancer is inversely related to sun exposure. These findings have epidemiologically linked prostate cancer to vitamin D metabolism.
As with breast and colon cancer, familial clustering of prostate cancer has been reported. Unlike with breast and colon cancer, however, specific genetic lesions have not been identified to merit the routine use of genetic screening for prostate cancer. The search for “prostate cancer genes” has identified candidate genetic events implicated in tumorigenesis, but these findings have been more useful in understanding the underlying etiology of prostate cancer than in screening. For example, a major hereditary prostate cancer susceptibility locus resides at 1q24, although the responsible gene(s) remains under investigation (19,20). More recently, men carrying BRCA1/BRCA2 (breast cancer 1 and 2) mutations have been shown to be at increased risk of developing prostate cancer (21,22). In addition, it has been suggested that BRCA-associated prostate cancer cases may be more virulent than nonfamilial cases (23,24). However, it remains unclear how this information will influence management decisions for individual patients.
RELEVANT HISTOMORPHOLOGY OF PROSTATE CANCER
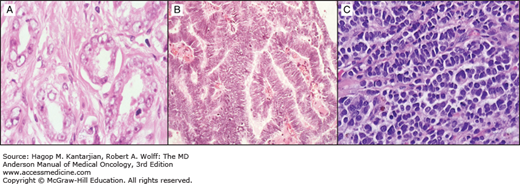
Most epithelial cancers arise from the prostatic acinus, with fewer than 10% having a pure ductal origin (this is the opposite of the pattern seen in cancers of the pancreas and breast, where ductal cancers are far more common than those arising in the acinar portion of the secretory unit). The majority show glandular differentiation (ie, adenocarcinoma) (Fig. 37-2A). Importantly, mucin is essentially never seen in prostate cancer. Historically, numerous grading systems have been devised, using all of the typical morphologic criteria by which pathologists can sometimes infer biologic potential. While more than 20 systems have been put forward, prostate cancers are now almost universally graded according to the system of Gleason.
The Gleason system is unique among pathologic grading systems because it is a composite classification based on a combination of architectural and cellular features often considered in the grading of other epithelial cancers. As originally described, the Gleason system includes two components:
There are five patterns, or grades, ranging from normal architecture to arrays of cells without any glandular organization at all. At our institution, because unequivocal criteria for malignancy already correspond to Gleason grade 2 and the Gleason grade 1 category is rarely reported, prostate cancers are assigned Gleason grades ranging from 3 through 5.
The Gleason score is obtained by assigning one Gleason grade to the most dominant (ie, “primary”) pattern and another to the next most common (ie, “secondary”) pattern. By convention, such data are expressed as a sum, with the primary pattern listed first. Thus, the Gleason score can range from 2 (ie, 1 + 1) to 10 (5 + 5). At MD Anderson, these scores range from 6 to 10, reflecting our decision not to assign a Gleason grade of 1.
A major advantage of the Gleason grading system is its reproducibility and ability to distinguish different cancer phenotypes that influence clinical management. The system is most useful for tumors falling at one the extremes (eg, Gleason 6 or less versus Gleason 8 to 10). However, a major disadvantage of the system is that it does not provide a refined view of Gleason 7 tumors, the most commonly reported type. Gleason 7 cancers (ie, 3 + 4 or 4 + 3) represent a clinically heterogeneous group with variable biologic potential and clinical outcome (25). Despite efforts to improve stratification of Gleason 7 tumors, it is clear the Gleason system is inherently limited by light microscopic methodology to evaluate morphology. Thus, most investigators are now looking to incorporate molecular characterization of tumors into the current grading system.
Gleason grading is a validated system to prognosticate untreated prostate cancers sampled at initial diagnosis. However, the common practice of applying Gleason grading to treated cancers may be misleading. At MD Anderson, for example, pathology reports of specimens obtained after hormone ablation typically indicate “hormonal treatment effect” rather than a Gleason grade per se. Thus, Gleason scores from serial biopsies obtained pre- and posttherapy from the same patient should be interpreted with careful regard to treatment effects. To address this problem, our group has proposed a novel “posttherapy” histologic classification to introduce uniformity in analysis of treated tissue specimens (26). If prospectively validated, this system should prove useful in prognosticating preoperatively treated prostate cancers.
An additional confounding variable is tissue sampling. We recognize that there is intratumoral heterogeneity of grade within individual prostate cancers. Thus, it logically follows that the extent and areas of sampling will affect the accuracy of grading and staging. The development of techniques to obtain prostate tissue samples with ultrasound guidance and limited patient morbidity has had a major effect on stage and grade assignment. We increasingly find that the completeness of tissue sampling as measured by the number and distribution of transrectal biopsies greatly influences the adequacy of tumor staging and grading. Biopsy algorithms have been developed to ensure adequate number and distribution of the biopsies (27).
Many of the challenges attributed to the sampling errors that occur with needle biopsies can be overcome with proper handling of the prostate following surgery. Assigning a primary and secondary Gleason score is straightforward in a properly processed specimen. This requires great attention to detail in assessing whether the cancer has invaded beyond the prostate capsule (extracapsular) or beyond the margin of resection (margin positive). Effective communication between the surgeon and pathologist is essential to properly identify the extent and site of surgical margin involvement. As the therapeutic benefit of postoperative radiation therapy is better appreciated, the importance of these aspects has proportionally increased.
In prostate cancer, gene fusions between TMPRSS2 (21q22.3), an androgen-regulated gene, and ERG, from the ETS family of transcription factors (21q22.2), are common (28). However, the functional and prognostic significance of TMRPSS2-ERG remain poorly understood. Currently, detection and measurement of TMPRSS2-ERG are not performed in standard clinical practice.
Additional studies have sought to refine the diagnostic classification of prostate cancer through molecular methods. Unfortunately, no consensus yet exists about the usefulness of molecular phenotypic (or genotypic) characterization in prostate cancer. Using immunohistochemistry, we do know that expression of specific proteins in human specimens has been linked to the clinical course of the disease. For example, loss of functional PTEN, mutations in p53, and increased expression of Bcl-2 are some of the widely reported gain- and loss-of-function changes that have been correlated with prostate cancer progression and mechanistically linked to castration-resistant growth (29,30). Despite this link to biology and correlation with clinical outcome, the methods provide insufficient additional clinical information to justify their routine use.
The two most prominent morphologic subtypes are ductal and small cell/anaplastic variants.
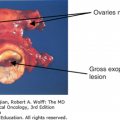
The ductal variant of prostate cancer in its pure form is unusual. More often, ductal and acinar components are mixed, and the relative contribution of the ductal variant to the clinical phenotype of the cancer is unclear. Our impression of the behavior of ductal cancers of the prostate is based on those tumors that are either dominant or pure ductal in origin (Fig. 37-2B). The clinical features that lead us to suspect its presence include the lack of proportional rises in serum PSA concentrations with invasion of the base of the bladder (occasionally mistaken for urothelial cancer), soft tissue distribution of metastases, or lytic bone metastases. For localized disease, patients with pure ductal adenocarcinoma have a better clinical outcome after radical prostatectomy than patients with mixed ductal adenocarcinoma (31). However, metastatic cancers as more aggressive and demonstrate a higher probability of developing visceral metastases than pure acinar adenocarcinomas.
Small cell carcinoma of the prostate is clinically distinguishable from prostate adenocarcinoma in predictable ways. It is castration resistant, is highly metastatic, produces little or no PSA, and causes lytic rather than blastic bone metastases. In addition, in comparison to adenocarcinoma, small cell prostate cancer more frequently metastasizes to lymph nodes and visceral organs (eg, liver or lung). It can arise de novo or more commonly as a delayed manifestation of progression in patients with a history of high-grade adenocarcinomas following therapy (hormone ablation, radiation therapy, or chemotherapy). The classic clinical presentation is a patient with a precipitously enlarging prostate associated with obstructive symptoms and very little (to no) PSA production. Interestingly, the evolution to a neuroendocrine phenotype is often associated with expression of carcinoembryonic antigen (CEA). For many patients, the CEA will be a much more useful monitoring tool than PSA.
In our experience, small cell carcinomas are also frequently detected in metastatic sites. Our approach is to biopsy sites with unusual features (lytic as opposed to blastic metastases or soft tissue metastases), particularly in those patients whose PSA concentration is judged to be lower than would be expected for the volume of cancer. Requests are made to pathology to analyze the tissue for neuroendocrine markers (chromogranin, synaptophysin, etc). Although standard criteria for establishing a diagnosis have not emerged, most of these cancers will show “salt-and-pepper” chromatin, express synaptophysin and chromogranin, and display a high nuclear/cytoplasm ratio (Fig. 37-2C), However, the diagnosis of small cell carcinoma does not require proof of neuroendocrine differentiation.
For this reason, we have added the term anaplastic to our nomenclature to describe aggressive tumors presenting clinically as “neuroendocrine-like” but lacking neuroendocrine markers. Until more precise genotype-phenotype associations are elucidated, we recognize that tumors displaying the clinical phenotype described may display heterogeneity with respect to neuroendocrine differentiation. Small cell/anaplastic carcinomas are highly responsive to etoposide and cisplatin-based chemotherapy but are generally incurable (32,33,34).
The search for premalignant lesions of the prostate has resulted in the identification of two candidate morphologic lesions (35). The first and most promising premalignant lesion is prostatic intraepithelial neoplasia (PIN). Grades I and II PIN are observed but have not been reliably associated with cancer or linked with confidence to prostate cancer development or progression. Thus, the reporting of grades I and II PIN has fallen into disfavor at MD Anderson and at most leading institutions. Grade III PIN has been linked to the presence of cancer in many studies, but it does not justify a therapeutic intervention (eg, prostatectomy). Rather, its presence leads to the recommendation of a more thorough biopsy to search for coexistent cancer or a repeat biopsy within 6 to 12 months. Grade III PIN is frequently linked to the presence of established cancer in other regions of the prostate, and as a consequence it rarely serves as a useful early predictive marker. Therein lies the difficulty of performing prevention studies targeting PIN. Most MD Anderson clinicians share the view that a report of multifocal PIN III is nearly identical to a report of low-grade prostate cancer, but with the important caveat that there is insufficient evidence to justify routine intervention (surgery or radiation).
The second potential premalignant lesion is proliferative inflammatory atrophy (PIA) (36). Histologically, these lesions are characterized by inflammatory infiltrates associated with atrophic epithelium. Compared with normal epithelium, there is an increased fraction of proliferating epithelial cells within PIAs. These lesions are thought to provide a mechanistic link between chronic infection or inflammation and the predisposition to develop prostate cancer. However, in contrast to PINs, adenocarcinomas rarely arise from PIAs, and PIAs are often observed in prostate biopsies that have no evidence of cancer. Thus, it remains unclear if PIAs are truly precursor lesions for prostate cancer. Ongoing studies seek to address this.
It is difficult to precisely assess the extent of prostate cancer on clinical criteria alone. The major benchmark of local extent that influences treatment—organ confined versus non–organ confined—is essentially impossible to distinguish by rectal examination and is not easily appreciated by any imaging modality. Furthermore, PSA levels do not accurately inform about extraprostatic extension of disease. Thus, it is common to employ an array of modalities, including transrectal ultrasound, computed tomography, conventional magnetic resonance imaging (MRI), MRI with an endorectal receiver coil, complex biopsy strategies designed to sample the seminal vesicles and extraprostatic space, and even pelvic lymph node sampling, to determine disease extent. Clearly, each of these modalities offers a different level of sensitivity for detecting lesions and can vary markedly in efficacy. In the final analysis, the concept of clinical stage is meaningless without proper context. One must ask, What is the clinical stage by a particular set of diagnostic tests¿
With the advent of PSA screening, there has been a dramatic increase in the number of younger men detected with localized disease. Along with improved outcomes for local therapy (surgery or radiation), it logically follows that PSA screening has contributed to the improved survival rates for men diagnosed with localized prostate cancer. However, the benefits of PSA screening remain controversial for clinicians and a source of confusion for patients. For clinicians, there has been a lack of level 1 evidence supporting the use of PSA screening. The two large randomized screening trials (with greater than 250,000 patients) has not helped clarify the issue because one trial did not show a survival benefit (the PLCO Cancer Screening Trial in the United States), while the other one did (the ERSPC trial in Europe) (37,38). While confounding factors to each study limit a definitive conclusion about PSA screening, the enormous expense and time required may discourage future PSA screening trials. Available evidence favors clinician discussion of the pros and cons of PSA screening with average-risk men aged 55 to 69 years. Other strategies to mitigate the potential harms of screening include considering biennial screening, a higher PSA threshold for biopsy, and conservative therapy for men receiving a new diagnosis of prostate cancer.
Although the US Preventive Services Task Force no longer recommends routine screening (39), in our practice, we recommend annual PSA and digital rectal examination (DRE) for all healthy men starting at age 50 or age 40 for those at high risk (eg, those men with a family history or African Americans). Early inclusion of the patient in the decision-making process is essential to optimize patient care given the ambiguities regarding the cost effectiveness and clinical value of widespread screening for prostate cancer. The rationale for routine screening that justifies routine application is outlined in Table 37-1.
|
Given the high prevalence of prostate cancer and the significant burden of therapy for patients, there is great interest in preventing the disease altogether or preventing lethal progression of the disease. Inhibitors of 5α-reductase enzymes (types 1 and 2 enzymes convert testosterone to dihydrotestosterone, the predominant and more potent agonist of AR signaling in prostate tissues) have demonstrated potential in this regard. Two randomized, placebo-controlled clinical trials utilizing finasteride (a type II inhibitor) and dutasteride (a type I/II inhibitor) have demonstrated a reduction in prostate cancer in healthy men who had no evidence for prostate cancer at enrollment but did have risk for developing disease (based on age and PSA) (40,41). A third clinical trial evaluated the ability of dutasteride to prevent prostate cancer progression in patients with low-grade, low-risk, localized prostate cancer at study entry (REDEEM), and dutasteride could provide a favorable addition to active surveillance for men with low-risk prostate cancer (42). While we have not adapted chemoprevention as standard of care, we are encouraging patients with low-risk prostate cancer to participate in preoperative trials that offer short-duration 5α-reductase inhibition prior to radical prostatectomy. Our goal is to elucidate molecular signatures that (1) predict response (or resistance) to 5α-reductase inhibition, (2) characterize therapy-specific effects on epithelial-stromal compartments, and (3) refine existing risk stratification schemas.
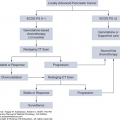
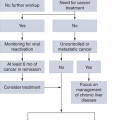
PATIENT MANAGEMENT BY DISEASE STATE
As a result of widespread screening of men by PSA and DRE, the majority of patients are seen with clinically localized disease at diagnosis. Unfortunately, it is not always obvious how to match the individual patient with the most appropriate management. In an abstract sense, patients with clinically localized prostate cancer fall into one of four theoretical categories:
Those not destined to have any clinical manifestations of their disease. These patients are actually harmed by any intervention, including further surveillance.
Those destined to have a clinical manifestation of cancer but will not to die of it. These patients might benefit from definitive therapy (such as prostatectomy or radiation) but would likely benefit equally from less-morbid intervention (eg, minimally invasive surgery).
Those destined to have life-threatening disease for whom definitive therapy will be curative. Patients who can be cured, or for whom there will be a substantial alteration of the natural history of their disease, constitute the group that will unequivocally benefit from definitive local therapy.
Those destined to have life-threatening disease for whom the opportunity to cure by means of local therapy either never existed or passed. For these patients, however, control of the primary could still be an important component of an overall treatment strategy that considers the probability of local versus distant progression, comorbidity, and other factors.
The common practice of urologists and radiation therapists is to assume that nearly every patient falls into the third category, and thus they recommend definitive therapy for the vast majority of newly diagnosed cases of localized prostate cancer. Unfortunately, available evidence suggests that less than half of patients are in category 3, so it is not surprising that understanding the role of definitive therapy in eliminating prostate cancer morbidity and mortality has been both difficult and controversial. In fact, these issues underscore the fact that overtreatment of “clinically insignificant” prostate cancers certainly occurs. The significant cost and morbidity associated with local treatment also adds to the difficulty of managing these patients (whether the patient ultimately benefited from the therapy or not).
In patients with newly diagnosed localized disease, current prognostic and predictive models rely on data derived from large prostatectomy series conducted at major academic centers. For example, investigators at Johns Hopkins initially published a predictive model relating the rate of finding disease that is not confined to the prostate (by assessing the surgical specimen) as a function of three readily available preoperative clinical parameters: PSA, Gleason score from the core biopsy, and the clinical stage based on DRE (43,44). The correlation of these features with pathologically organ-confined disease, summarized in the famous “Partin tables,” provided sobering evidence that commonly encountered subsets of patients had a surprisingly high risk of disease that was not confined to the prostate. Of course, not all patients with pathologically organ-confined disease relapse, and not all patients with pathologically organ-confined cancers are cured. Thus, the importance of this particular surrogate outcome was and remains uncertain. Nevertheless, the effect of the Partin tables on clinical practice has been profound. They have driven the application of prostatectomy to patients with smaller and smaller volumes of cancer. It is clear that although more patients are remaining free of disease after prostatectomy, this comes paradoxically at the cost of operating on many patients who may not have needed surgery or not operating on many patients who would have benefited from good local control even if the surgery were not curative.
Additional models have been developed to predict outcomes following radical prostatectomy or radiation therapy. Based on the work of D’Amico, a combination of pretherapy PSA, Gleason score, and clinical stage can be used to stratify patients into low (T1-T2a and Gleason score 2-6 and PSA <10 ng/mL); intermediate (T2b-T2c or Gleason score 7 or PSA 10-20 ng/mL); high (T3a or Gleason score 8-10 or PSA >20 ng/mL); and locally advanced (T3b-T4) groups that predict risk for both biochemical recurrence and survival following definitive local therapy (radical prostatectomy or radiation) (45). Similarly, Kattan et al developed postoperative nomograms for predicting prostate cancer recurrence after radical prostatectomy (46). These tools not only help guide recommendations for individual patients, but also help stratify patients for clinical trials. For example, low-risk patients can be directed toward “active surveillance” trials, while high-risk patients can be direct toward adjuvant/neoadjuvant trials. The rationale for the use of predictive nomograms is outlined in Table 37-2.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


