Fig. 6.1
Intrahepatic assembly of apoB into VLDL
VLDL assembly and secretion is a process highly regulated by the availability of triglycerides in the liver [22]; TG may derive from different sources, including uptake of albumin-bound fatty acids, uptake of circulating remnants of VLDL and chylomicrons, and de novo hepatic synthesis. Beside apoB and MTP, TG availability determines the efficiency of apoB-lipoprotein formation. In fact, reduced lipid availability results in targeting of apoB for degradation and decreased VLDL secretion [23]. Fatty acids derived from diet or released from adipose tissue enter the liver where they are re-esterified, forming triglyceride droplets [24]. Not all mobilized TGs enter into the secretory pool to contribute to VLDL formation; a relatively large proportion (determined by MTP and insulin activity) is returned back to the cytosolic pool. Newly synthesized VLDL contain little apoC; after secretion in the circulation, they acquire apoE and additional apoC from HDL (Fig. 6.2).
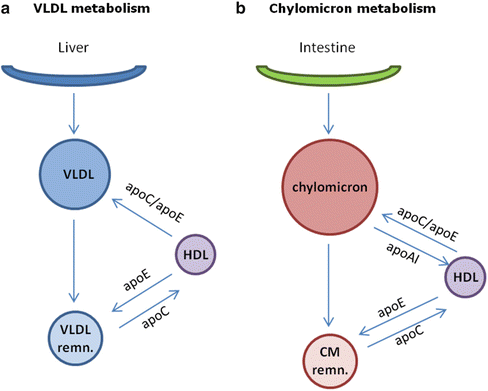
Fig. 6.2
Metabolism of VLDL (a) and chylomicrons (b)
Two major subfractions of VLDL exist, large VLDL1 and smaller VLDL2. VLDL1 secretion is dependent on fatty acid availability, and insulin inhibits VLDL1 secretion [25]; this does not seem to be true for VLDL2. After secretion, VLDL1 are delipidated, following hydrolysis of TG through lipoprotein lipase; the delipidation process of VLDL1 is not complete, and only a minor fraction is converted to LDL, most remnants being directly removed from plasma [26]. On the contrary, most VLDL2 particles are delipidated and converted to LDL [27].
Chylomicron Assembly and Secretion
Three proteins play a key role in the process of chylomicron assembly: apoB48, MTP, and apoA-IV. ApoB48 is produced from the same gene of apoB100 in the small intestine and is formed by post-transcriptional mRNA editing in intestinal enterocytes; it lacks the LDLR-binding domain and is essential for the assembly of chylomicrons. MTP is a lipid-transfer protein that transports ER membrane-bound lipid (mainly TG) to newly synthesized apoB48, a step that prevents apoprotein degradation; moreover, MTP facilitates the successive lipidation of chylomicron precursors. ApoA-IV is a lipid-binding protein expressed mainly in the small intestine and incorporated early into nascent chylomicrons; after chylomicron secretion, apoA-IV dissociates from the particles to circulate as lipid-free protein.
Chylomicrons are responsible for the transport of dietary cholesterol and medium- and long-chain fatty acids from the intestinal lumen to the liver. The main lipids in chylomicrons are triacylglycerols. They are assembled mainly in the ER and then transported to the Golgi via specialized vesicles (PCTVs, prechylomicron transport vesicles). During the first assembly step, apoB48, synthesized by the small intestine, is translated into the ER lumen and immediately lipidated through the action of intestinal MTP (Fig. 6.1), resulting in the formation of a precursor particle. The lipidation can occur both by transfer of lipid from the ER membrane to apoB48 or by binding of MTP to apoB48 to facilitate the protein folding and lipid acquisition. During the second step, MTP mediates further addition of lipids to the precursor. In this phase apoA-IV is added at the particle surface; apoA-IV increases MTP activity and increases chylomicron lipidation [28].
Lipoprotein Lipase-Mediated Lipolysis
VLDL and chylomicrons leave the liver and intestine and enter the circulation where they acquire apoC-II and apoE from plasma HDL. In the capillaries of adipose tissue and muscle, triacylglycerols are hydrolyzed by endothelial lipoprotein lipase (LPL, activated by apoC-II) to produce free fatty acids which are then absorbed by the tissues. During the removal of fatty acids, a large percentage of the phospholipids and apoproteins are transferred to HDL, converting the lipoproteins to VLDL and chylomicron remnants (Fig. 6.2).
Hepatic Clearance of Remnants
The main organ involved in the clearance of remnant lipoprotein is the liver, where hepatocytes express LDL receptor (LDLR), LDL receptor-related protein 1 (LRP1), and heparan sulfate proteoglycans (HSPGs) in high amounts. In concert with LPL and hepatic lipase (HL), these surface molecules facilitate the rapid hepatic clearance of remnant lipoproteins [29–32] that are extremely atherogenic [33] (Fig. 6.3). The most critical molecule in the remnant clearance is apoE, involved in the binding of lipoprotein to the LDLR family and HSPGs [31]. Multiple steps are involved in the uptake of remnants by hepatocytes. HSPGs interact with apoE present on the remnant surface and sequester them in the space of Disse [32]; moreover, HSPGs can bind LPL and HL that eventually may continue their lipolytic activity and prepare the particles for the successive internalization process [32, 34], which is mediated by LDLR, HSPGs, and the HSPGs/LRP complex.
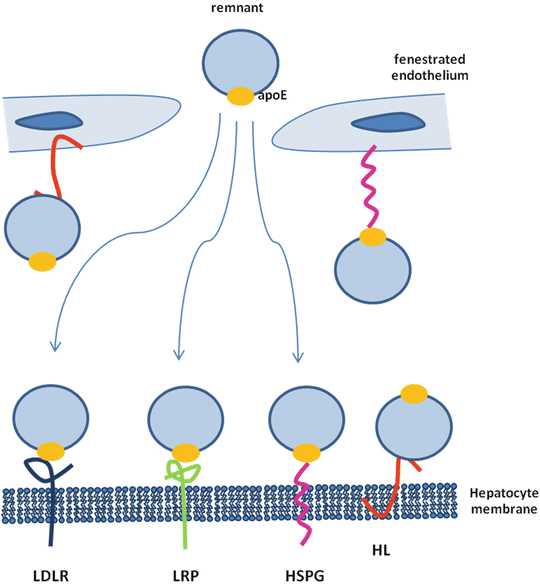
Fig. 6.3
Pathways of hepatic clearance of remnants
Chylomicron remnants contain mainly cholesteryl esters, apoE, and apoB48 and return to the liver where they are taken up by hepatocytes via interaction with the LDL receptor which requires apoE [35]. Moreover, chylomicron remnants can acquire additional apoE, allowing the remnants to be taken up via the chylomicron remnant receptor, a member of the LDL receptor-related protein (LRP) family [35]. Alternatively, chylomicron remnants can remain sequestered in the space of Disse by binding of apoE to heparan sulfate proteoglycans and/or binding of apoB48 to hepatic lipase [35]. During this phase, chylomicron remnants may be further metabolized which increases apoE and lysophospholipid content, allowing for transfer to LDL receptors or LRP for hepatic uptake. VLDL remnant particles are immediately cleared by the liver or, alternatively, further modified by HL and cholesteryl ester transfer protein (CETP) to generate LDL.
The Role of Insulin in TGRLs Metabolism
The VLDL assembly process in the liver is tightly regulated by insulin [36, 37]: under fasting conditions, VLDL production in the liver is induced; on the contrary, in response to post-prandial insulin release, hepatic VLDL production is repressed [25, 38, 39]. This tight regulation allows the liver to rapidly adapt to metabolic shifts between fasting and feeding and to maintain plasma lipids within the physiological range [25, 39, 40] (Fig. 6.4).
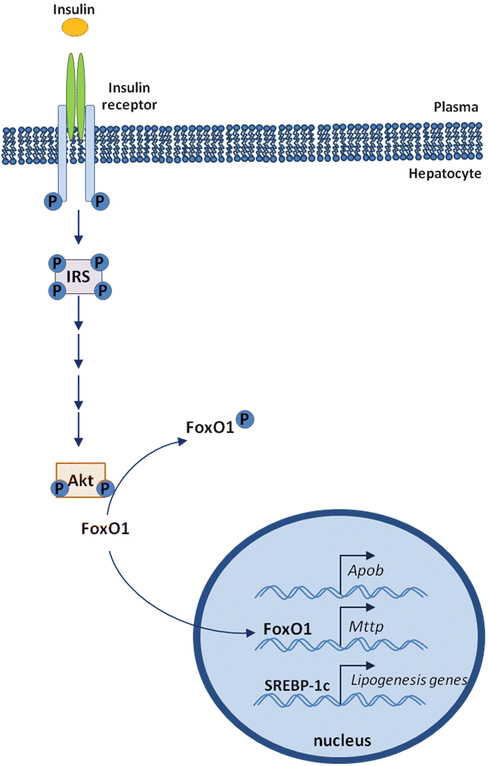
Fig. 6.4
Insulin regulation of FoxO1 activity
Several observations suggest that insulin inhibits apoB secretion by inducing its degradation [41–44]; alternatively, insulin reduced free fatty acid (FFA) availability by reducing FFA mobilization from adipose tissue, resulting in apoB secretion inhibition [45]. The apoB gene is believed to be constitutively expressed as hepatic mRNA levels in vivo tend to be stable in most animal systems. However, several studies suggest that apoB mRNA abundance can be influenced by insulin in vivo [46, 47]. Hepatic apoB production is mainly regulated at the post-translational level by lipid availability, a process that is inhibited by insulin, resulting in an acute inhibitory effect of insulin on hepatic VLDL-triglyceride secretion to limit postprandial plasma lipid excursion. Hepatic apoB mRNA is stimulated by forkhead box O1 (FoxO1) and inhibited by insulin in a cell system [48]; moreover, hepatic activity of FoxO1 is increased during fasting and inhibited in response to feeding [49]. These observations suggest an additional mechanism by which the liver controls hepatic apoB production at the transcriptional level.
In the liver, insulin acts on fatty acids similarly to glucose: it promotes the storage of glucose as glycogen, and fatty acids as triglycerides during feeding. This will result in decreased hepatic VLDL secretion and decreased hepatic glucose release. Moreover, a decreased VLDL secretion during feeding limits the increase of plasma triglycerides during the prandial phase, when intestinal fats are absorbed to produce chylomicrons, which in turn deliver fatty acids to adipose tissue.
FoxO1 (forkhead box O1) is a transcription factor that plays a role in regulating hepatic glucose metabolism during fasting by inducing the expression of genes involved in gluconeogenesis [50]. In addition, FoxO1 may regulate lipid metabolism by inducing hepatic MTP expression, resulting in increased secretion of VLDL [48]. Under physiological conditions, this effect is reversed by insulin [48]. In the absence of insulin, FoxO1 is localized in the nucleus in a transcriptionally active form and induces the expression of MTP; after insulin release, FoxO1 is phosphorylated and translocated out of the nucleus, resulting in inhibition of FoxO1 transcriptional activity [51] (Fig. 6.4).
Insulin Resistance
Diabetes is characterized by hyperglycemia due to either defects in insulin secretion and/or insulin properties. Patients with insulin resistance are at high risk of developing diabetes and cardiovascular (CV) disease [52]. Insulin resistance is a condition of reduced responsiveness of tissues (liver, muscle, and adipose tissue) to normal circulating levels of insulin [53, 54], a condition present in different diseases, including type 2 diabetes [55], obesity, hypertension, and dyslipidemia [56]. As a result, insulin production increases to maintain normal levels of blood glucose. Insulin is a hormone essential for the maintenance of glucose homeostasis, secreted by the pancreatic β-cells mainly in response to increased circulating glucose levels after a meal [57].
When the concentration of blood glucose increases, the pancreas releases insulin into the circulation. In muscle and adipose tissues, insulin binds to cell surface receptors [58]. Following this binding, several biochemical signals are activated within the cells to take up glucose and convert it to energy [59]. If the pancreas fails to produce enough insulin or the insulin receptors do not function properly, the cells cannot uptake glucose and the level of glucose in the blood remains high.
Several defects can determine insulin resistance, including insulin receptor defects, insulin signaling defects [59, 60], mutations in insulin signaling molecules [61], and mitochondrial dysfunction [60]. In the early stages of insulin resistance, the pancreas compensates by producing more insulin to control the increased levels of glucose in the blood. This results in high blood glucose levels and high blood insulin levels (a condition known as hyperinsulinemia) at the same time. If this condition is not treated, the islets of Langerhans (the insulin-secreting groups of cells) in the pancreas may eventually shut down and decrease in number. When an insulin-resistant subject cannot maintain the degree of hyperinsulinemia required to bypass the defective action of insulin, type 2 diabetes develops.
The Role of Insulin Resistance in TGRLs Metabolism
In animal models of insulin resistance, hepatic MTP mRNA levels are significantly higher with simultaneous increase in VLDL levels [62–64]; finally, treatments that ameliorate insulin resistance and dyslipidemia determined reduced MTP expression and VLDL levels [49, 65, 66]. These observations suggest that in insulin-resistant subjects, MTP expression is no longer regulated by insulin, resulting in VLDL overproduction.
Insulin resistance is considered mainly a carbohydrate metabolism disorder; however, lipid and lipoprotein abnormalities are observed in individuals with insulin resistance [67]. These abnormalities include (1) increased plasma levels of VLDL triglyceride and apoB100, (2) reduced plasma levels of HDL and apoA-I, and (3) relatively normal LDL levels with increase of small dense LDL particles.
Acute insulin infusion reduces production of TG-rich VLDL in healthy non-obese humans [25, 40, 68, 69]; this effect can result from several mechanisms, including inhibition of hepatic MTP expression [70], increased apoB degradation [43], and inhibition of VLDL particle maturation [71]. This suppressive effect of insulin is, however, attenuated or even reversed [41, 72] when exposure to insulin is prolonged (such as in conditions of insulin resistance [68, 69]), where an increase in VLDL (mainly in the VLDL1 fraction) production is observed [73–75]. These observations suggest that chronic hyperinsulinemia plays a role in mediating the increased production of hepatic VLDL. In addition, insulin resistance of adipose tissue increases the levels of circulating free fatty acids that can enter into the liver, thus stimulating VLDL production [76]. Finally, loss of insulin inhibition of FoxO1 activity in insulin resistance increases the production of both glucose and VLDL-TG, contributing to the dual pathogenesis of hyperglycemia and hypertriglyceridemia in diabetes.
Hepatic TG in Insulin Resistance
Fatty acid flux to the liver is increased in insulin resistance [77, 78], due to a failure of insulin to inhibit TG lipolysis in adipose tissue [79]. Increased levels of fasting and post-prandial TG are features of insulin resistance [80]. The increase in post-prandial TG is due both to defective lipolysis of VLDL and chylomicrons, combined with increased VLDL secretion [80], and to increased production of chylomicrons [81]. In addition, insulin resistance also reduces lipoprotein lipase activity secondary to increased apoC-III (an inhibitor of LPL) secretion [82], resulting in reduced lipolysis of VLDL and chylomicron TG.
Another source of hepatic TG is de novo lipogenesis that contributes significantly to VLDL lipidation and production in insulin-resistant subjects. The main transcription factor of de novo lipogenesis is SREBP-1c (sterol response element-binding protein-1c) [83] that in turn is regulated by LXR (liver X receptor) [84]. Insulin plays a key role in the expression of hepatic SREBP-1c, in part by stimulating LXR expression [85, 86]; furthermore, insulin promotes the maturation of SREBP-1c independently of LXR [87].
Intestinal lipoprotein production is increased in insulin resistance; chylomicron overproduction is in fact a consequence of impaired insulin regulation. Under physiological conditions, chylomicron production is inhibited by insulin; this inhibitory process is lost or reduced in the presence of impaired insulin responsiveness. Increased postprandial TG was thought to be due to reduced chylomicron and VLDL lipolysis, combined with increased VLDL secretion [80, 88]. However, increased assembly and secretion of apoB48-containing chylomicrons has been observed in hyperinsulinemic conditions [81]. The elevation of free fatty acids in plasma increases not only hepatic, but also intestinal, lipoprotein production [81, 89], suggesting that the intestine responds to insulin resistance similarly to the liver [90].
Diabetes and Hepatic Uptake of Remnant Lipoproteins
Diabetes impairs hepatic uptake of remnant lipoproteins [29, 91, 92]. Under these pathological conditions, LDLR does not seem to significantly contribute to the reduced uptake of remnant lipoprotein [93, 94]. The major contribution to this effect appears to be related to HSPGs; as they are not proteins, a high number of genes involved in their assembly and disassembly must be regulated, both at translational and at posttranslational levels [95, 96]. In type 1 diabetes, hepatic HSPGs exhibit sulfation defects [97, 98], due to the suppression of a crucial enzyme in HSPG assembly [96]; moreover, the farnesoid X receptor, a known inducer of HSPG expression [99], is suppressed [100]. In type 2 diabetes and other diseases characterized by insulin resistance, proteoglycans exhibit several defects, including decreased sulfation [101, 102]. Insulin resistance also induces the hepatic overexpression of the heparan sulfate glucosamine 6-O-endosulfatase-2 (SULF2), an enzyme that degrades cell surface and matrix HSPGs, thus reducing the catabolism of remnant lipoprotein and contributing to postprandial dyslipoproteinemia in type 2 diabetes [103].
Triglyceride-Rich Lipoproteins and Vascular Dysfunction
The elevation in circulating free fatty acids impairs endothelium-dependent vasodilatation [104], and the decreased endothelial function may be dependent on enhanced oxidative stress [105]. The changes induced by TGRLs in the post-prandial phase are even more deleterious in terms of endothelial dysfunction and inflammation; indeed, several in vivo studies have demonstrated that post-prandial hypertriglyceridemia impairs endothelial function [106, 107]. Post-prandial hypertriglyceridemia is also associated with an inflammatory state and enhanced levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, soluble intercellular adhesion molecule (sICAM)-1, and soluble vascular cell adhesion molecule (sVCAM)-1 [108–110].
The molecular mechanisms underlying these effects of TGRLs have only recently been studied in detail. There are a few key issues that should be taken into account when analyzing the vascular effects of TGRLs. TGRLs derive either from an exogenous pathway (chylomicrons and chylomicron remnants containing apolipoprotein apoB48) or from a liver-derived pathway (VLDL and VLDL remnants containing apoB100). Under fasting conditions, however, chylomicrons are rapidly metabolized, thus the TGRL fraction is mainly composed of apoB100-rich particles, and the remnants derive mainly from the catabolism of VLDL (small VLDL and intermediate-density lipoprotein). In several dyslipidemic conditions, chylomicrons are metabolized at a lower rate, resulting in the accumulation of chylomicron remnants in the fasting state. In the post-prandial state, an enormous production of TGRLs containing both apoB48 and apoB100 occurs, leading to an impaired endothelial function. This dysfunction rapidly fades away in normotriglyceridemic subjects where the TGRLs are efficiently metabolized, whereas the condition persists in hypertriglyceridemic patients where TGRLs accumulate in the circulation. TGRLs undergo lipolysis mediated by lipoprotein lipase (LPL), generating different biologically active products that may affect endothelial cell function [111].
Early studies using HepG2 cells investigated the intracellular signaling pathway induced by VLDL exposure [112]. VLDL-induced protein kinase C activity results in the activation of mitogen-activated protein kinase (MAPK). Studies conducted in endothelial cells (ECs) indicate that VLDL can also activate nuclear factor (NF)-κB [113], a transcription factor that plays an important role in the phenotypic modulation of ECs in a pro-inflammatory condition. To date, plasminogen activator inhibitor-1 is the only gene that has been shown to be consistently induced in ECs to a larger extent when comparing VLDL from patients with hypertriglyceridemic type IV and type II versus VLDL from normolipidemic subjects [114]. Both in human umbilical vein ECs and human aortic ECs, TGRLs from hypertriglyceridemic subjects induce an increased mRNA expression of adhesion molecules, such as VCAM-1, platelet/endothelial cell adhesion molecule (PECAM)-1, and endothelial/leukocyte adhesion molecule (ELAM)-1, while TGRLs from normolipidemics induced VCAM-1 expression in both the cell lines and ELAM-1 selectively in the aortic ECs, but to a lesser extent [115]. Specific inhibition of p38 mitogen-activated protein kinase and NF-κB suggests a major involvement of these factors in adhesion molecule expression induced by TGRLs in both NTG and HTG patients. Furthermore, TGRLs induced monocyte chemoattractant protein (MCP)-1 expression in ECs, suggesting that activation of the endothelium by TGRLs could support both adhesion and transmigration of leukocytes. In addition, TGRLs from hypertriglyceridemic patients induced IL-6 expression. Again, these effects are mainly dependent on NF-κΒ activation.
The composition of the TGRL particles plays a key role in determining the pro-inflammatory response to TGRLs [116]. A different composition of VLDL (fatty acid, lipids, and apoproteins) may be responsible for the differences observed between normolipidemic and hypertriglyceridemic TGRLs. TGRLs isolated following a meal enriched in saturated fatty acids induced E-selectin and VCAM-1 expression to a higher extent than TGRLs isolated after a meal enriched in monounsaturated and polyunsaturated fatty acids [116]. Furthermore, lipolysis products from TGRLs increase endothelial permeability, perturb zonula occludens-1 and F-actin, and induce apoptosis [111]. One could speculate that, in the presence of hypertriglyceridemia, the reduced activity of LPL may promote the presence of pro-inflammatory TGRLs.
Although hypertriglyceridemia is an independent risk factor for coronary artery disease [117], accumulating evidence suggests that post-prandial (hyper)lipidemia contributes to the development of atherosclerosis and coronary artery disease [118]. Several studies have demonstrated that postprandial hypertriglyceridemia impairs endothelial function, suggesting a role for triglycerides in the initiation and further progression of atherosclerosis [106, 107]. Post-prandial hypertriglyceridemia is associated with an inflammatory state and enhanced levels of TNF-α, IL-6, sICAM-1, and sVCAM-1 [108–110]. Although TGRLs isolated from fasting plasma samples of hypertriglyceridemic subjects induce an inflammatory response in ECs [115], ECs incubated with post-prandial TGRL demonstrated an increased mRNA expression of VCAM-1, ELAM-1, P-selectin, PECAM-1, and ICAM-1. Similarly, post-prandial TGRLs increased ICAM-1 and VCAM-1 protein expression [119]. Also fasting TGRLs increase adhesion molecule expression, however, the effect observed with post-prandial TGRL is much more pronounced. Furthermore, ICAM-1 expression was induced solely upon incubation with post-prandial TGRLs. Likewise, MCP-1 and IL-6 expression was induced upon incubation with post-prandial TGRLs; again, this effect is more pronounced than that observed with fasting TGRLs. As the induction of adhesion molecules and the increased release of cytokines and chemokines have been associated with endothelial dysfunction [120], our data suggest that endothelial activation by TGRL occurs during the post-prandial phase and may promote the endothelial dysfunction observed after a meal. Notably, a single high-fat meal led to a significant elevation of endothelial microparticles, known to be a sensitive indicator of endothelial disturbance, in healthy normolipidemic subjects [107]. This observation suggests that endothelial microparticles may be an indirect marker of endothelial dysfunction or injury induced by postprandial TGRL.
TGRLs and their remnants are present within human and experimental atherosclerotic lesions [121–123]: chylomicron remnants directly penetrate the endothelial cell layer and are entrapped within the subendothelial space, leading to focal accumulation [122] (Fig. 6.5). TGRLs may directly contribute to the atherosclerotic process by inducing endothelial dysfunction [124], by enhancing monocyte adhesion [125], and by triggering lipid accumulation within the artery wall [126]. Exposure to TGRLs, especially those isolated from patients with type 2 diabetes [127], leads to the intracellular accumulation of triglyceride and/or cholesteryl ester in human monocyte- [127] and murine-derived macrophages [126, 128]. Abnormal reverse cholesterol transport and low levels of high-density lipoprotein associated with hypertriglyceridemia [129, 130] can accelerate the lipid deposition process within arterial macrophages. The interaction of TGRLs with cholesterol-loaded human macrophages increases the cell lipid content while compromising the subsequent efflux of cholesterol to lipid-poor apoA-I [131]. These aspects may contribute significantly to the generation of macrophage foam cells in vivo and might account for the accelerated atherogenesis observed in patients with type 2 diabetes. Finally, remnant lipoproteins induce smooth muscle cell activation and proliferation [132, 133].
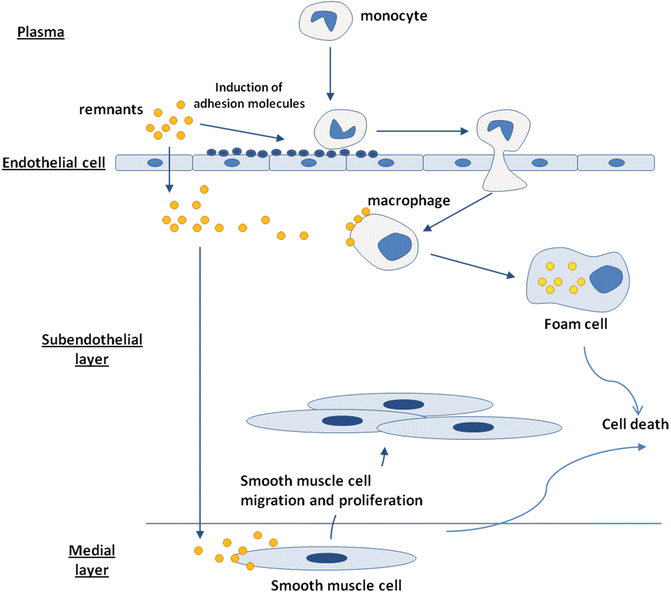
Fig. 6.5
Remnant contribution to the atherosclerotic lesion development
Most of the available evidence suggests that in normolipidemic subjects either in the fasting state or in the post-prandial phase, TGRL may affect endothelial function only when a pro-inflammatory environment is already present and may perhaps contribute to accelerating the damage induced by other lipid and non-lipid factors. However, in hypertrygliceridemic patients, TGRLs from the fasting state and postprandial phase can both induce endothelial dysfunction by promoting a pro-inflammatory activation of the endothelium. These findings are in line with the idea that these lipoproteins may play a significant role in the early stages of atherogenesis.
References
1.
Karpe F, Bell M, Bjorkegren J, Hamsten A. Quantification of postprandial triglyceride-rich lipoproteins in healthy men by retinyl ester labeling and simultaneous measurement of apolipoproteins B-48 and B-100. Arterioscler Thromb Vasc Biol. 1995;15: 199–207.PubMed
2.
Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46: 733–49.PubMed
3.
Chen SH, Habib G, Yang CY, et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238:363–6.PubMed
Related posts:
 Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus
Roles of Extravasated and Modified Plasma Lipoproteins in Diabetic Retinopathy
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus
Roles of Extravasated and Modified Plasma Lipoproteins in Diabetic Retinopathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


