Fig. 3.1
Insulin resistance (IR) is selective to the PI3K (predominantly metabolic) effects of insulin. Hyperinsulinemia, a common concomitant of IR, results in exaggerated MAPK (predominantly nuclear and mitogenic) effects of insulin
Atherosclerosis
Ross and Glomset [21] proposed more than three decades ago a proliferative model for atherosclerosis, where endothelial denuding injury led to platelet aggregation, release of platelet-derived growth factor, and proliferation of smooth muscle cells in the arterial intima, thereby forming the nidus of the atherosclerotic plaque and updating the centuries’ old Virchow’s concept of “response to injury” model (initially proposed in 1856) which envisaged atherosclerosis merely as a passive deposition of lipid debris in arterial walls. This simplistic concept has since evolved largely due to the advances in cell biology techniques, and current thinking is that atherosclerosis is indeed a complex process invoking endothelial dysfunction, vascular smooth muscle dysfunction, immune dysfunction, and inflammation [22, 23].
Atherosclerosis is an inflammatory process that selectively affects arteries and is highly prevalent in both genders. Thrombo-occlusive complications of atherosclerosis including stroke and myocardial infarction are major causes of morbidity and mortality. Thrombo-occlusive complications of atherosclerosis including stroke and myocardial infarction are major causes of morbidity and mortality. Atherosclerosis is perhaps initiated by endothelial dysfunction and in the presence of structural alterations such as the absence of a confluent luminal elastin layer and the exposure of proteoglycans [2], in which apolipoprotein-B (apoB) enriched particles such as low-density lipoprotein (LDL) accumulate in the subendothelial space. Elevated levels of circulating LDL cholesterol (LDL-c) facilitate atherosclerosis and cardiovascular disease (CVD) [24]. ApoB100 binding to negatively charged extracellular matrix proteoglycans leads to intimal retention of LDL particles, where they are vulnerable to oxidative modification by reactive oxygen species and enzymes such as myeloperoxidase or lipoxygenases released from inflammatory cells. Oxidized LDL (oxLDL) promotes expression of adhesion molecules and the secretion of chemokines by endothelial cells, which in conjunction with platelet-derived chemokines drive immune cell infiltration into the intima. Early lesions (“fatty streaks”) consist of T cells and monocyte-derived macrophage-like foam cells loaded with lipids. Accrual of dying cells and other cellular debris along with cholesterol crystals forms a necrotic core. Fibroatheromatous plaques are covered by a fibrous cap composed of collagen and smooth muscle cells (SMCs), which are replaced by macrophages in the thinning inflamed caps that are prone to rupture. The “shoulder” regions are heavily infiltrated by T cells and mast cells, which produce enzymes and proinflammatory mediators, contributing to adventitial inflammation of advanced plaques [25]. The pathogenesis of atherosclerosis is discussed in greater detail elsewhere [26].
IR is associated with processes that facilitate atherosclerosis (see Fig. 3.2). The pathophysiological processes involved in the initiation and progression of early atherosclerotic lesions are somewhat different from those associated with the formation of clinically dangerous plaques [27, 28], and distinguishing the effects of IR and hyperglycemia on these processes is important. As alluded to earlier, early-to-mid-stage atherogenesis involves the subendothelial retention of apoB-containing lipoproteins, activation of endothelial cells, recruitment of monocytes and other inflammatory cells, cholesterol loading of lesional cells, and migration of smooth muscle cells to the intima. In contrast, advanced plaque progression is influenced primarily by processes that promote plaque necrosis and thinning of a collagenous “scar” overlying the lesion, called the fibrous cap. The objective of this chapter is to describe how IR and hyperglycemia promote atherogenesis and plaque progression. It should be noted that IR and hyperglycemia are likely to have additive or synergistic pro-atherogenic effects in the setting of T2DM. For example, glucotoxicity may contribute to IR, and treatment of hyperglycemia in T2DM has been shown to improve IR in some tissues [29].
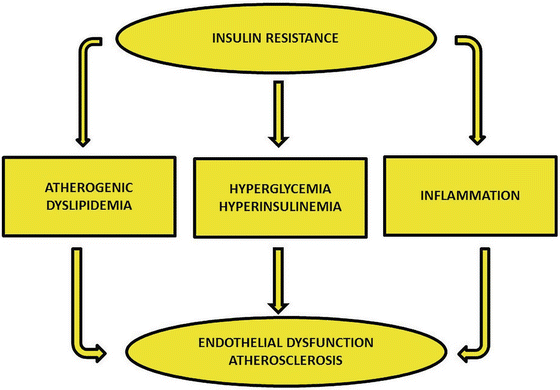
Fig. 3.2
Insulin resistance-related processes are associated with endothelial dysfunction and promotion of atherosclerosis
Role of Atherogenic Dyslipidemia
Altered metabolism of triglyceride-rich lipoproteins (TGRLP) is an important part of the metabolic environment in insulin-resistant states. Figure 3.3 illustrates the lipid pathways that operate in IR states. Insulin promotes synthesis of nonesterified fatty acids (NEFA) or free fatty acid (FFA) and its assimilation into triacylglycerols. In obesity and other IR states, excessive adipose tissue breakdown leads to increased hepatic delivery of FFA. This leads to increased hepatic synthesis and secretion of triglyceride-laden very low-density lipoprotein (VLDL). Impaired clearance of VLDL and chylomicrons (intestinally derived) leads to prolonged plasma retention of these particles. These partially lipolyzed remnants, which include cholesterol-enriched intermediate-density lipoproteins (IDLs), are very atherogenic in humans and in a number of animal models [30, 31]. Increased hepatic production and/or slow plasma clearance of VLDL leads to increased production of precursors of small dense LDL (sdLDL) particles. As many as seven distinct LDL subspecies which differ in their metabolic behavior and pathological roles have been identified [32]. Plasma VLDL levels correlate with increased density and decreased size of LDL [33, 34]. In addition, LDL size and density are inversely related to plasma levels of high-density lipoprotein (HDL), especially the HDL2 subclass [35]. sdLDL particles arise from the intravascular processing of specific larger VLDL precursors in multiple steps, including lipolysis [32]. TG enrichment of the lipolytic products occurs mainly through the action of cholesteryl ester transfer protein (CETP) and, together with hydrolysis of triglyceride and phospholipids by hepatic lipase, leads to increased production of sdLDL particles [30, 31]. Another reason for prolonged plasma retention time for these particles is the reduced affinity of the hepatic LDL receptors for these particles [32].
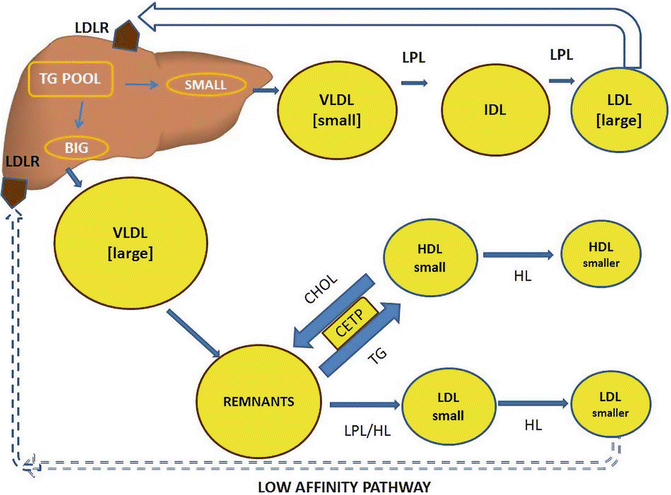
Fig. 3.3
Insulin resistance: lipid pathways leading to atherogenic dyslipidemia
HDL particles are heterogeneous, and multiple subclasses differing in diameter and density have been identified, ranging from the small dense HDL3c, HDL3b, and HDL3a to the larger HDL2a and HDL2b [36]. The reasons for the reduction in plasma HDL concentration in IR states are multifactorial, but a major factor appears to be the increased transfer of cholesterol from HDL to TGRLP, with reciprocal transfer of triglyceride to HDL. Triglyceride-laden HDL particles are hydrolyzed by hepatic lipase and subsequently rapidly catabolized and cleared from plasma [37]. Typically, the reduced HDL levels in plasma of patients with T2DM are manifest as reductions in the HDL2b subspecies and relative or absolute increases in smaller denser HDL3b and HDL3c.
Increased atherogenicity of sdLDL appears to be related to a number of properties, including reduced LDL receptor affinity [38, 39], greater propensity for transport into the subendothelial space [40], increased binding to arterial wall proteoglycans [41], and susceptibility to oxidative modifications [42–44]. These in vitro findings corroborate the concept that sdLDL contributes to arterial damage in patients with the atherogenic dyslipidemia seen in insulin-resistant states.
Dysglycemia and Atherosclerosis: Possible Mechanisms
Epidemiological research suggests the presence of an association between glycemic control and CVD risk [45]. Data from the United Kingdom Prospective Diabetes Study (UKPDS) suggested more or less a linear relationship between hemoglobin A1c (HbA1c) and CVD end points, particularly myocardial infarction [46]. However, the association between HbA1c and microvascular complications is stronger than it is for macrovascular outcomes such as myocardial infarction, stroke, or cardiovascular death, raising the question if glucose plays a greater role in the pathogenesis of microvascular than macrovascular outcomes in DM. Similar relationships between hyperglycemia and CVD outcomes have been demonstrated in T1DM, but the association seems less pronounced [47].
It has been suggested that glucose might act directly or indirectly via the generation of advanced glycation end products (AGEs) or reactive oxygen species. AGEs are a class of chemical by-products that result from the combination of protein and sugar (usually glucose) and are increasingly recognized as a mediator of hyperglycemia-induced cytopathology. Hyperglycemia inside the cell increases diacylglycerol (DAG) levels, a critical activating cofactor for the classic isoforms of protein kinase C, kinase-β, kinase-δ, and kinase-α [48–51]. Protein kinase C (PKC) activation leads to a variety of gene effects. The vasodilator nitric oxide (NO) levels are low because nitric oxide synthase (eNOS) expression is reduced, while the vasoconstrictor endothelin-1 expression is increased. Levels of transforming growth factor-β (TGF-β) and plasminogen activator inhibitor-1 (PAI-1) are also increased [51–55]. Several studies have demonstrated that inhibition of PKC prevented early renal and retinal complications of DM [52, 55, 56]. PKC activation has been linked to increased inflammation via increased nuclear factor κB (NFκB) activation [57, 58], which in turn leads to the expression of several proinflammatory genes, including adhesion molecules that facilitate monocyte adhesion to endothelial cells [57]. This eventually leads to foam cell formation. Glucose has also been shown to affect monocyte/macrophage activation in vitro. Monocytes exposed to high glucose concentration show increased expression of interleukin-1β (IL-1β) and interleukin-6 (IL-6) [59]. This results in induction of PKC, activation of NFκB, and robust release of superoxide, which could play a role in glucose-mediated oxidative stress [59]. Glucose auto-oxidation results in the generation of several reactive oxygen species such as superoxide anion, which can facilitate LDL oxidation in vitro [60]. Cell-surface scavenger receptors on arterial macrophages take up modified lipoproteins including oxidized LDL (oxLDL) that have become oxidized as a result of glucose-mediated oxidative stress [60, 61] or modified by AGEs [61]. Moreover, AGE-modified albumin can inhibit scavenger receptor B1 (SR-B1)-mediated efflux of cholesterol to HDL [62]. AGE proteins in the circulation may also interfere with the functions of SR-B1 in reverse cholesterol transport by inhibiting the selective uptake of HDL-cholesteryl ester, as well as cholesterol efflux from peripheral cells to HDL. Thus, hyperglycemia affects alterations in the delivery and removal of lipid from macrophages by lipoproteins and other proteins.
Endothelial Dysfunction in Insulin-Resistant States
Studies in human obesity and IR have revealed a clear association between the chronic activation of proinflammatory signaling pathways and decreased insulin sensitivity. For example, elevated levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-8 (IL-8) have all been reported in various diabetic and insulin-resistant states [63–67]. The inflammatory marker C-reactive protein (CRP), a nonspecific acute-phase reactant synthesized predominantly in the liver, is commonly elevated in states characterized by IR [68]. Also, experiments in naturally occurring rodent models of obesity, knockout, and transgenic mice as well as detailed studies of insulin signaling at the molecular level have begun to elucidate the mechanistic links between obesity-induced inflammation and insulin-resistant states.
The precise pathways by which accrual of excess adipose tissue initiates systemic inflammation are unclear. One theory holds that adipose tissue expansion leads to adipocyte hypertrophy and hyperplasia, eventually outstripping the local oxygen supply leading to hypoxia and activation of cellular stress pathways [69]. This causes cell-autonomous inflammation and the release of cytokines and other proinflammatory signals. Adipokines such as resistin, leptin, and adiponectin perhaps also affect inflammation and insulin sensitivity. Locally secreted chemokines attract proinflammatory macrophages into the adipose tissue where they form crown-like structures around large dead or dying adipocytes. These tissue macrophages then release cytokines that further perpetuate inflammation involving neighboring adipocytes, thereby exacerbating inflammation and IR. Hepatic inflammation from steatosis occurs in obesity, whereby hepatocyte stress pathway responses may be triggered resulting in hepatocyte-autonomous inflammation. Activation of Kupffer cells (macrophage-like cells in the liver) releases locally acting cytokines and exacerbates inflammation and promotes hepatic insulin resistance. In addition, overnutrition is often accompanied by elevations in tissue and circulating FFA concentrations, and saturated FFAs can directly activate proinflammatory responses in vascular endothelial cells, adipocytes, and myeloid-derived cells [70]. A state of systemic inflammation then occurs.
TNF-α, a proinflammatory cytokine secreted predominantly by monocytes and macrophages, has varied biological effects on lipid metabolism, coagulation, and endothelial function. Activation of the TNF receptor results in the stimulation of NFκB signaling via inhibitor of nuclear factor kappa-B kinase subunit beta (IKKb). A landmark study by Hotamisligil [71] showed that adipose tissue isolated from different obese rodent models overexpressed TNF-α. His group also showed that neutralization of TNF-α in obese fa/fa rats ameliorated IR [71]. Similar correlations between TNF-α levels, obesity, and IR were soon demonstrated in humans [67]. Corresponding in vitro experiments demonstrated that by activating IKKb, TNF-α stimulation leads to serine phosphorylation of Irs, attenuating its ability to transduce insulin-mediated cellular events [72]. Mice genetically deficient in TNF-α or the TNF-α receptor 1 gene (TNFR1) do not develop IR caused by high-fat feeding or obesity [73].
TNF-α also affects insulin signaling independent of Irs1. TNF-α-treated cultured 3T3-L1 adipocytes show reduced expression of the insulin receptor, Irs1, and Glut4 genes, as well as a decrease in insulin-stimulated glucose uptake [74]. Ruan et al. also showed that TNF-α induced a decrease in many 3T3-L1 adipocyte genes, including GLUT4, hormone-sensitive lipase (HSL), long-chain fatty acyl CoA synthetase, adiponectin, the transcription factor CCAAT-/enhancer-binding protein-alpha (C/EBP), and the nuclear receptors peroxisome proliferator-activated receptor gamma (PPARɣ) and retinoic acid x receptor (RXR). As many of these genes have direct and indirect effects on glucose homeostasis, changes in adipocyte expression of these genes will likely contribute to IR [75].
c-Jun N-terminal kinase 1 (JNK1) (encoded by MAPK8) also contributes to the development of IR in obese and diabetic states. Hirosumi et al. [76] found elevated JNK activity in liver, adipose tissue, and skeletal muscle of obese insulin-resistant mice, and knockout of JNK1 (JNK1−/−) resulted in the amelioration of IR in high-fat-diet-fed (HFDF) mice. At the cellular level, these workers also showed that JNK1 knockout led to decreased Irs1 phospho-Ser307 in the liver. Importantly, deletion of JNK1 also caused resistance to the development of obesity, so the improved insulin sensitivity in these animals could be a result of decreased adiposity and/or decreased JNK1 activity in insulin target cells. The role of JNK2 has also been assessed in studies and seems to play a significant role in the development of obesity-induced IR. Recent data suggest that JNK2 can be involved in metabolic regulation when JNK1 is absent, since JNK1 +/− JNK2 −/− mice phenocopy JNK1 −/− mice in their reduced adiposity and improved insulin sensitivity [77]. It appears that functional in vivo interactions between these isoforms may contribute to the regulation of insulin action.
Salicylate and its derivative acetyl salicylic acid (or aspirin) have been in use to treat symptoms of T2DM for a very long time. At higher doses, they are effective in reducing blood sugar levels, but adverse effects such as gastrointestinal bleeding and tinnitus have precluded their widespread use in this context [78]. These agents are weak inhibitors of IKKb, thus preventing Irs1 serine307 phosphorylation with some insulin-sensitizing effects [79]. Kim et al. [80] showed that lipid infusion causes acute IR in rodents and pretreatment of lipid-infused rats with salicylates improves glucose utilization in skeletal muscle, as measured during hyperinsulinemic-euglycemic clamp studies. They also performed lipid infusions in IKKb heterozygous knockout mice (IKKb+/−) and reported similar improvements in insulin sensitivity when compared to wild-type controls [80]. Yuan et al. showed that TNF-α treatment of 3T3-L1 adipocytes induces IR, an effect that could be prevented by pretreatment of cells with aspirin. A parallel experiment was performed in adipocytes using okadaic acid to activate IKKb independent of TNF-α stimulation, and again, aspirin prevented okadaic acid-induced IR. In vivo studies of aspirin-treated obese rats and mice have shown that salicylate pretreatment protects them from IR [81]. Mice with a liver-specific constitutively active IKKb transgene (LIKK) developed hyperglycemia and decreased hepatic insulin sensitivity with mild secondary systemic IR in skeletal muscle. There was liver expression of the proinflammatory markers IL-6, IL-1β, and TNF-α similar to that found in the liver of obese mice. In rescue experiments, LIKK mice treated with sodium salicylate or IL-6-neutralizing antibodies had markedly improved insulin sensitivity. In addition, mice expressing the liver-specific IκBα super-repressor transgene (LISR), which prevents the activation of IKKb, protected both LIKK and obese mice from hepatic IR. Another important finding in this study was the elevated expression of the macrophage-specific markers, Emr1 (also known as F4/80) and Cd68, in the livers of LIKK and obese mice. Co-expression of LISR and LIKK in compound transgenic mice reduced both IR and the expression of these same macrophage markers. These data indicate that hepatic inflammation caused by a high-fat diet is mediated by both hepatocytes and Kupffer cells (liver macrophages) [82]. These studies highlight the role of IKKb in the development of obesity and inflammation-induced IR.
As alluded to before, nitric oxide (NO) is an endogenous signaling molecule produced by nitric oxide synthase and acts as a signal transduction molecule for a number of physiological processes such as vasodilation. It is also involved in many pathophysiologic states such as IR. Several IR inducers such as FFAs, proinflammatory cytokines, and oxidative stress activate the expression of Nos2, the gene that encodes iNOS [83]. NO reduces Akt activity by causing s-nitrosylation of a specific cysteine residue [84]. Increased iNOS activity also results in the degradation of Irs1 in cultured skeletal muscle cells [83]. Nos2 knockout mice are protected from obesity-induced skeletal muscle IR, and this is associated with improved PI3K-Akt activity [85]. It appears that Nos2 is also required for the development of sepsis-induced skeletal muscle IR, perhaps also mediated by the s-nitrosylation of the insulin receptor Irs1 and Akt [86, 87]. These studies suggest that increased iNOS activity may play a direct role in the pathogenesis of IR.
Macrophages and lymphocytes elaborate interleukin-10 (IL-10) which exerts anti-inflammatory activity by inhibiting TNF-α-induced NFκB expression, via reduction in IKK activity and inhibition of NFκB DNA-binding activity [88]. It has been shown in human subjects that IR is more prevalent in subjects with reduced serum levels of IL-10 [89]. Consistent with the concept that IL-10 may have insulin-sensitizing effects is the laboratory evidence that mice treated with IL-10 did not become insulin resistant when treated with either IL-6 or lipid infusions [89]. Lumeng et al. showed that IL-10-treated 3T3-L1 adipocytes are protected from TNF-α-induced cellular IR [90]. Recombinant IL-10 therapy of conditions such as psoriasis has raised hopes that immunomodulation of IL-10 activity can be a potential treatment of IR [91].
Atherosclerosis in Insulin-Resistant States: Role of Macrophage
A key discovery in the arena of obesity-induced inflammation and IR was the finding that bone marrow-derived macrophages are present in adipose tissue of obese mice and humans [92, 93]. Weisberg et al. compared adipose tissue RNA profiles for various mouse models of obesity and found that a subset of genes, while not typically expressed in adipocytes, were confirmed through immunohistochemistry to be adipose tissue-resident macrophage-derived. The percentage of macrophages in a given adipose tissue depot positively correlated with adiposity and adipocyte size. They also found that adipose tissue macrophages were responsible for nearly all adipose tissue TNF-α expression and a significant portion of Nos2 and IL-6 expression. They quantified the infiltration of macrophages in subcutaneous adipose tissue from obese human subjects and reported that as high as 50 % of the total cell content consists of macrophages compared to 10 % in lean controls [92]. Xu et al. [93] reported similar findings and showed that thiazolidinedione (TZD) treatment could repress the expression of macrophage-specific genes, providing an additional mechanism by which TZD treatment improves insulin sensitivity.
A study by Arkan et al. [94] showed that inhibition of the macrophage inflammatory pathway protects mice from obesity-induced insulin resistance. In this study, the investigators generated both a myeloid-specific deletion of IKKb (IKKbΔmye ) and liver-specific deletion of IKKb (IKKbΔhep ). They found that IKKbΔhep mice are protected from high-fat-diet-induced hepatic IR but that this was a tissue-autonomous effect, since these mice still developed IR in the muscle and fat. There was a significant reduction in the expression of inflammatory markers in the liver suggesting that inactivation of inflammation can prevent HFD-induced insulin resistance. Also, tissue-specific deletion of IKKb in myeloid cells (IKKbΔmye mice) led to improvement in insulin sensitivity with globally improved insulin action in the muscle, liver, and fat. As such, these results showed that inactivation of myeloid-IKKb activity prevented systemic IR, most likely by blocking local paracrine interaction between resident macrophages and insulin target tissues.
Monocyte chemoattractant protein-1 (MCP-1), also known as chemokine ligand 2 (Ccl2), and its cognate receptor chemokine receptor 2 (Ccr2) are also major components of IR in obese mice. MCP-1 is a chemokine secreted primarily by macrophages and endothelial cells that promotes the recruitment of monocytes to inflamed tissues. Ccr2 is expressed in monocytes but also in the lung, spleen, and thymus [95]. Weisberg et al. found that obesity-matched Ccr2 −/− mice displayed reduced adipose tissue macrophage infiltration, reduced hepatic steatosis, decreased inflammatory profiles, and improved systemic insulin sensitivity. Ccr2 deficiency also attenuated high-fat-diet-induced weight gain by causing a reduction in caloric intake, highlighting the possible involvement of Ccr2 in the control of eating behavior. Also, treatment of obese mice with a pharmacological antagonist of Ccr2 led to decreased adipose tissue macrophage infiltration and improved insulin sensitivity [96]. Comple-mentary studies on MCP-1 have shown that its expression is increased in obese mice, suggesting that changes in MCP-1 levels promote the recruitment of macrophages to adipose tissue which then causes inflammation and IR. Studies on transgenic mice that overexpress MCP-1 under the control of the adipose tissue-specific AP2 promoter found that MCP-1 overexpression is associated with macrophage infiltration and IR [97, 98]. Kanda et al. [98] also showed that the onset of these abnormalities in obese mice could be prevented by genetic deletion of MCP-1. MCP-1 may also have a role in energy metabolism. Unlike other studies, Inouye et al. [99] showed that HFD-fed MCP-1 KO mice developed hyperinsulinemia and increased adiposity independent of adipose tissue macrophage levels, which were unchanged. Differences in experimental approaches as well as the complexity/redundancy of chemokine signaling may have accounted for these conflicting conclusions. In total, most evidence suggests that the MCP-1/Ccr2 axis could provide an important mechanistic link between obesity, adipose tissue inflammation, and IR.
Related posts:
 Laboratory Assessment of Lipoproteins in Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Lipoproteins and Diabetic Nephropathy
The PPAR System in Diabetes
Laboratory Assessment of Lipoproteins in Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Lipoproteins and Diabetic Nephropathy
The PPAR System in Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


