Fig. 4.1
Lipoprotein structure involves amphipathic surface molecules PL and UC and the core consists of a variable mixture of nonpolar CE and TG. Providing structure and solubility are surface apolipoproteins. Because the volume of a sphere is related to the third power of the radius, even small lipoproteins can traffic significant numbers of lipid molecules per particle
Table 4.1
Human apolipoproteins
Apolipoprotein | Molecular weight | Lipoprotein association | Function |
|---|---|---|---|
ApoA-I | 28,331 | HDL, chylomicrons | Activates ACAT, ABCA1 |
ApoA-II | 17,380 | HDL, VLDL, Chylos | FFA metabolism, RCT, antiox |
ApoA-IV | 44,000 | Chylomicrons, HDL | Chylo production, RCT, LCAT |
ApoA-V | 39,000 | Chylos, VLDLs, HDLs | TG metabolism |
ApoB-48 | 240,000 | Chylomicrons | Structural protein |
ApoB-100 | 513,000 | VLDL, IDL, LDL | Binds to LDL receptor |
ApoC-I | 7,000 | VLDL, HDL | Inhibits C-11, CETP |
ApoC-II | 8,837 | Chylos, VLDL, HDL | Activates lipoprotein lipase |
ApoC-III | 8,751 | Chylos, VLDL, HDL | Inhibits lipoprotein lipase |
ApoD | 32,500 | HDL | CETP |
ApoE | 34,145 | Chylos, VLDL, IDL, HDL | Binds to LDLr and LRP |
Table 4.2
Lipoprotein properties
Density kg/L | Composition by weight (% by weight) | Relative volumea | Apolipoproteins | ||||
|---|---|---|---|---|---|---|---|
UC + CE | TG | PL | Protein | ||||
Chylomicron | < 0.95 | −5 | −90 | −4 | −1 | 700,000 | 8-48, A-1, C-1, C-11, C-111, E |
VLDL | <1.006 | 25 | 55 | 18 | 8 | 360 | 8-100, A-ll, C-1, C-11, C-111, E |
IDL | 1.006–1.019 | Between a VLDL and LDL | 8-100, E | ||||
LDL | 1.019–1.063 | 55 | 6 | 20 | −20 | 32 | 8-100 |
HDL2 | 1.063–1.125 | 22 | 5 | 33 | 40 | 3 | A-1, A-ll, C-1, C-11, C-111, E |
HDL3 | 1.125–1.210 | 17 | 3 | 25 | 55 | 1 | A-1, A-ll |
Lp(a) | 1.04–1.13 | 48 | 9 | 21 | 22 | − LDL | B-100, a |
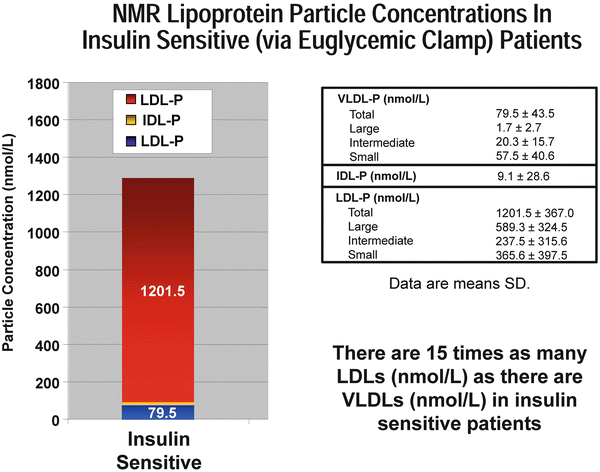
Fig. 4.2
NMR lipoprotein particle concentrations in insulin sensitive (via euglycemic clamp) patients. Adapted from Garvey et al. Diabetes 2003;52(2):453-462. Reference [110] in chapter
The apoA-I family of lipoproteins are the HDL class and they remain the most complex and enigmatic of all lipoproteins. HDLs not only traffic cholesterol and TG, but a large variety of other lipids including fat-soluble vitamins, phospholipids (e.g., phosphatidylcholine), and sphingolipids (sphingosin-1-phosphate, sphingomyelin, and ceramide), all of which are associated with many biological functions [14]. Collectively these are referred to as its lipidome. The protein cargo (proteome) of HDL is comprised of over 100 proteins, and these play defining roles in determining the functionality of HDL. HDLs acquire sterols from cells as they efflux UC, esterify the acquired UC using lecithin–cholesterol acyltransferase (LCAT), and then deliver the UC and CE elsewhere. HDLs are in a constant and dynamic state of lipidation and delipidation or remodeling and utilize a variety of cell membrane sterol transporters, lipid transfer proteins, and lipolytic enzymes to accomplish such [15]. There are many genetic, lifestyle, hormonal, metabolic, and inflammatory influences on HDL’s makeup and function, and many of these may be manipulated by pharmacologic and lifestyle interventions. Structurally HDLs are similar to all lipoproteins with a one-molecule thick surface of PL and UC and a core of mostly CE but also a small amount of TG. The main structural protein of HDL is one to five copies of apolipoprotein A-I arranged in a “trefoil” configuration [10] (Fig. 4.3).
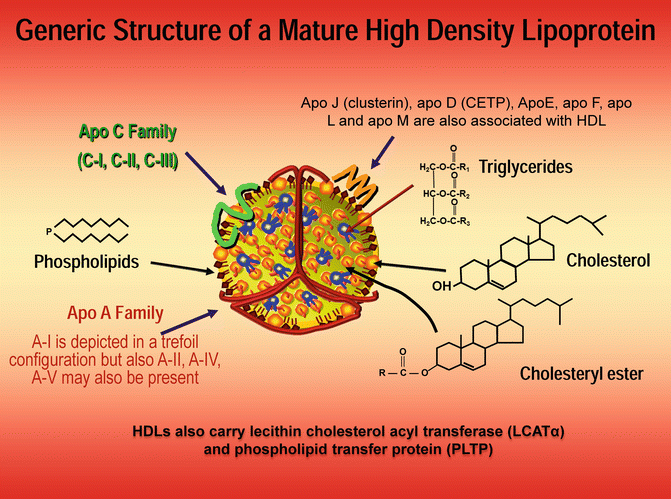
Fig. 4.3
HDL particles are very small lipoproteins with a core of TG and cholesterol ester. The major surface apoproteins are A-I, A-II, or A-IV and ApoC family and ApoE family. HDL-C refers to the cholesterol mass
HDL nomenclature can be very confusing and some terms are technology dependent. There is a numerical ultracentrifuge classification where super large HDLs (not always present) are called HDL1. As the particles shrink in size, the names change to HDL2b, HDL2a (both large with b being larger than a) and HDL3a, HDL3b and HDL3c (with 3a being the largest and 3c the smallest). These terms are also used by labs utilizing ultracentrifugation or gradient gel electrophoretic fractionation. NMR spectroscopic classification of HDLs uses the terms H1 through 5 (with 5 being largest). Labs using 2D electrophoresis with apoA-I staining report apoA-I, prebeta-HDL, and the α-HDL subspecies. Some laboratories report how much cholesterol is in various HDL subspecies (i.e., HDL2-C). The newest attempt from a group of experts to simplify HDL naming to simply refer to the particles as very small, small, medium, large, and very large. Classically those species have been called HDL1 (very large); HDL2a and HDL2b (large); HDL3a, HDL3b, and HDL3c (medium or small); and unlipidated apoA-I or prebeta (discoidal). Another system differentiates unlipidated apoA-I and prebeta HDLs (1 and 2) from large alpha HDLs (1, 2, 3, and 4 ranging from very large to small). NMR cannot measure unlipidated apoA-I or prebeta HDLs, but because of their transient existence, they only represent about 5 % of total HDL particles (HDL-P) [16] (Fig. 4.4).
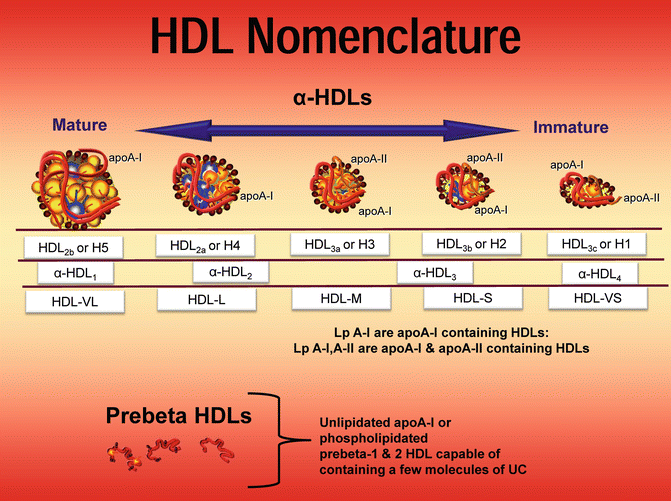
Fig. 4.4
HDL particles can be separated two-dimensional electrophoresis and apoA-I staining into prebeta and alpha lipoproteins. The former are unlipidated apoA-I or phospholipidated A-I. Each HDL can have from 1 to 5 molecules of apoA-I. The α-HDLs range in size from small and dense to larger and more buoyant. There can be from one to five molecules of apoA-I per HDL particle; thus, apoA-I is only an approximation of the number (concentration) of HDL particles. ApoA-II is present, predominantly on the smaller species. Dayspring original artwork
Measurement of Lipids and Lipoproteins
There are many ways of measuring lipoproteins in the laboratory including analyzing their density, their surface apolipoproteins, their core lipid content (expressed as TG or cholesterol mass per deciliter of plasma), or by NMR spectroscopy which determines the number of terminal methyl groups on CE, TG, and PL and translates that to particle numbers [17]. In clinical practice the majority of clinicians evaluate lipoproteins using lipid concentrations such as total particle cholesterol or TG or subparticle cholesterol mass per unit of plasma volume [18]. With respect to lab nomenclature and lipoprotein particle concentrations, (P) is added to the particle abbreviation and the value is expressed as nanomoles (nm) or micromoles (μmols) per liter. Lipoprotein particle concentrations can be determined using apolipoproteins, nuclear NMR [19], ion mobility transfer technologies [20], or ultracentrifugation with LDL staining.
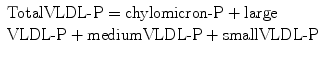
Total IDL-P (not typically separated into subparticles)
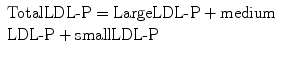
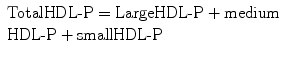
With respect to cholesterol mass measurements, (-C) is added to the particle abbreviation and the value is expressed as mg per deciliter (dL) or millimoles per liter (mmol/L).
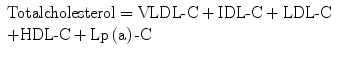
Calculated VLDL-C = TG/5 using the Friedewald formula [21].
Calculated 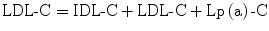 using the formula
using the formula ![$$\text{LDL}\text{-}\text{C}=\text{TC}\text{-}\left[\text{HDL}\text{-}\text{C}+\text{VLDL}\text{-}\text{C}\right]$$](/wp-content/uploads/2017/04/A314682_1_En_4_Chapter_IEq00042.gif) .
.
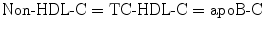
using the formula .One should never confuse specific lipoprotein lipid and lipoprotein concentrations such as LDL-C with LDL-P or apoB or HDL-C with HDL-P. Each is a valid way of measuring LDL or HDL and when they correlate highly (r values) with each other as they often do, they are said to be concordant. However, when cholesterol and lipoprotein particle concentrations do not correlate, they are said to be discordant. With respect to LDLs and HDLs, it is not uncommon to have high or low LDL-C and HDL-C with respective low or high LDL-P and HDL-P values. Lipoproteins that are CE-poor will require larger numbers of particles to traffic a given amount of core cholesterol and, conversely, cholesterol-rich LDL or HDL will require fewer particles to traffic the cholesterol mass. The cholesterol mass or number of cholesterol molecules per particle is a function of both the particle volume and core lipid makeup. Since the volume of a sphere is 4/3π(r 2), even subtle changes in particle diameter can cause tremendous changes in the number of particles required for lipid trafficking. The same is true of the particle core ratio of TG to CE [22, 23]. Adding to potential discordance between cholesterol mass and particle concentrations is the fact that both calculated and directly assayed cholesterol concentrations often fail to meet accuracy standards [24, 25].
Cellular Lipid Homeostasis
Because cholesterol can crystallize and cause cytotoxicity, cells maintain tight cholesterol homeostasis. All cells can synthesize UC as well as acquire it and FA through a variety of methods including cell membrane receptors which act as sterol or FA influx transporters or lipoprotein delipidation or internalization receptors (Table 4.3). Cells can also efflux sterols via free diffusion down their concentration gradient, a family of sterol efflux proteins called ATP-binding cassette transporters (ABC) [26], and UC, CE, TG, and PL can be exported by lipoprotein synthesis and secretion. Specialized cells such as enterocytes and hepatocytes can acquire UC through a sterol influx protein called the Niemann–Pick C1 like-1 protein (NPC1L1) which is expressed at both the jejunal lumen/enterocyte and the hepatobiliary interfaces [27]. Many cells express the scavenger receptor B1 (SR-B1) a bidirectional transporter which can participate in the efflux or influx of CE [28]. Sterols can also be effluxed from enterocytes and hepatocytes into the gut lumen or bile, respectively, using heterodimers of the sterol efflux transporters ABCG5 and ABCG8 [29]. Cells can also acquire UC and CE via receptors that internalize lipoproteins such as LDL receptors (LDLr) [30], LDL receptor-related proteins (LRP) [31], or ectopic β-chain of ATP synthase [32]. There are also putative receptors yet to be classified that perform these functions including an enterocyte protein involved with transintestinal cholesterol efflux (TICE) [33]. Cellular sterol homeostasis is regulated through synchronized action of all of the above mechanisms.
Table 4.3
Synthesis, remodeling, and catabolism of circulating HDL particles
Cell surface membrane receptors | |
|---|---|
Lipoprotein endocytosis | LDL receptor (LDLr) |
LDL receptor-related protein (LRP) | |
ApoA-1 beta-chain synthase or holoparticle receptor | |
Influx transporters | |
Niemann–Pick C1 like-1 protein (NPC1L1) | |
Scavenger receptor 81 (SR-81) | |
Fatty acid transport proteins (FATP) efflux transporters | |
ATP-binding cassette transporters; A8CA1, A8CG1, A8CG, A8CG8 | |
Scavenger receptor 81 | |
Putative transintestinal cholesterol efflux transporter | |
With respect to lipids, the human diet includes TG, FA, UC, CE, and phytosterols and to a lesser degree some stanols. Intestinal esterolases and lipases convert some of the ingested CE into UC and TG to FA and monoacylglycerols. However, after a meal the majority of the UC in the jejunum is of biliary origin. All of the lipids in the gut lumen are collectively organized and emulsified by lecithin (e.g., phosphatidylcholine) which is a phospholipid in biliary secretions. The lipids are then surrounded and organized by amphipathic bile acids into mixed biliary micelles which consist of mixtures of UC, phytosterols, stanols, phospholipids, monoacylglycerols, and FA. The micelles “ferry” these lipids to the epithelium of the intestinal microvilli. Once there, FA are absorbed into enterocytes by passive diffusion or fatty acid transport proteins [34]. The unesterified sterols, but not stanols, in the micelles are taken up by enterocytes via a sterol permease NPC1L1 protein, which utilizes other proteins (AP2-clathrin) to facilitate sterol absorption [35]. NPC1L1 is not involved with FA absorption and in part is regulated by PPAR-α and PPAR-Δ and is expressed at both the brush border of the intestinal epithelium and at the hepatobiliary cell junction [36]. Most humans absorb about 50 % of the sterols in the gut, but some people are hyperabsorbers (60–80 %) and some are hypoabsorbers (~20–40 %) [37]. NPC1L1 is also expressed at the hepatobiliary interface where it facilitates reentry of biliary UC back into the liver. Only UC, not esterified sterols, can be absorbed by NPC1L1. Once UC enters an enterocyte or hepatocyte, it is subject to esterification catalyzed by ACAT2 (acetyl-coenzyme A acetyltransferase 2) or within lipoproteins catalyzed by LCAT (lecithin–cholesteryl acyltransferase). Unlike UC, phytosterols are poor substrates for human ACAT and LCAT. Thus, ACAT2 in the enterocyte is a major regulator of sterol absorption [38]. Upon esterification of UC (the active and amphipathic form of cholesterol) becomes CE (the storage or transportation, hydrophobic form of cholesterol). In hepatocytes UC can upon exposure to 7α-hydroxylase be converted into the primary bile acids (cholate or chenodeoxycholate) which are effluxed into the biliary system via the bile salt export protein (ABCB11) [39].
The Apolipoprotein B Family of Lipoproteins
In the endoplasmic reticulum of enterocytes and hepatocytes, CE via the action of microsomal TG transfer protein joins with TG and apolipoprotein B48 (enterocyte) or B100 (hepatocyte) to form primordial chylomicrons or VLDL, respectively. Phospholipidation and additional TG lipidation of the particle occurs at the Golgi apparatus resulting in mature TG-rich lipoproteins. Evidence suggests apolipoprotein A-V (apoA-V) may have an inhibitory effect on this process as apoA-V modulates VLDL TG mobilization as well as secretion [40]. Also involved as a regulator of chylomicron synthesis and lipid absorption is apolipoprotein A-IV (apoA-IV), which, because of its large size, functions as a stabilizing, expandable lipid interface, enhancing particle formation. Interestingly, through effects on the hypothalamus and vagus nerve (gastric), apoA-IV also serves as a mediator of satiety and appetite [41].
Under physiologic conditions the largest lipoproteins, chylomicrons and VLDLs, traffic large amounts of TG and PL, which are released during TG-hydrolysis (de-esterification) or lipolysis. Under fasting conditions Friedewald noted almost all of the plasma TG are trafficked within VLDL particles and a typical VLDL carries five times more TG than cholesterol and thus VLDL-C can be estimated by dividing TG/5 [21] (Fig. 4.5). That calculation changed the practice of clinical lipidology as it allowed clinicians to calculate LDL-C using the formula LDL-C = TC − [HDL-C + VLDL-C]. If one assumes a normal TG value is < 150 mg/dL, then a desirable VLDL-C is ≤ 30 mg/dL [42]. An American Heart Association expert panel recently defined an optimal TG to be < 100 mg/dL [43]. As hydrolysis of TG occurs during lipase-mediated lipolysis, the TG-rich lipoproteins shrink and shed much of their surface PL which are picked up by PLTP (phospholipid transfer protein) and delivered to cells or maturing HDL particles. As the large TG-rich VLDL loses its core and surface lipids, it becomes smaller and denser. An LDL is basically a VLDL that has lost its TG and is therefore a cholesterol-enriched lipoprotein with a core of four or more times CE than TG. Any LDL, independent of its size, that has an excess core TG will be necessarily CE-poor [44]. Normally HDLs traffic very little TG and their core is 90–95 % CE; hence, TG-rich HDLs will be cholesterol-poor which can cause a low HDL-C value. The apoB-containing lipoproteins acquire their core lipids during their genesis in enterocytes or hepatocytes, whereas the apoA-I particles are sterol lipidated via a variety of cell membrane efflux transporters.
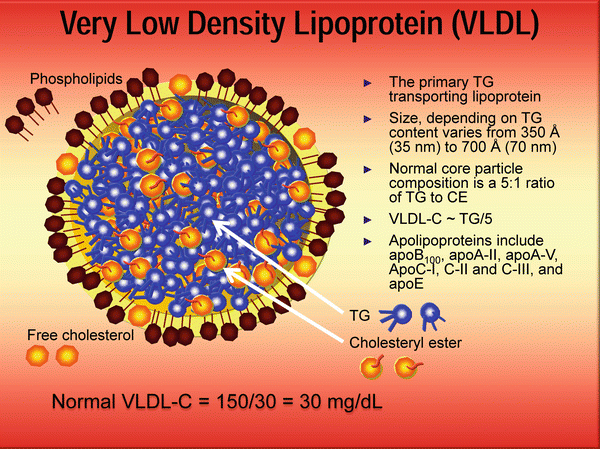
Fig. 4.5
Very-low-density lipoprotein is a large TG-rich particle that carries several apoproteins: ApoB100, ApoA-II, apoA-V, ApoC-I, apoC-II and apoC-III, and apoE. The primary function of VLDL is to traffic TG to myocytes and adipocytes and PL to the periphery. Dayspring artwork
Lipoproteins can also undergo additional lipidation or delipidation using a lipid transfer protein called apolipoprotein D (apoD) or cholesteryl ester transfer protein (CETP), which can exchange or swap one molecule of CE for CE, TG for TG, or CE for TG (often referred to as neutral lipids as they do not carry any charged group) [45]. The transfer of lipids between members of the apoB family themselves or the apoA-I family themselves is called a homotypic exchange, whereas the swapping of lipids between apoB and apoA-I particles is termed heterotypic. This lipid exchange is crucial to efficient lipid trafficking and dynamic remodeling of lipoproteins. Any lipoprotein acquiring TG will be subject to the lipolytic action of numerous lipases and, thus, TG-rich LDLs and HDLs would tend to become smaller and denser.
Other than apoB and apoA-I, there are numerous other apolipoproteins present on lipoproteins which perform multiple functions [46] (Fig. 4.6). All apoproteins but apoB are exchangeable, meaning they can transfer between lipoprotein species. Some act as ligands that direct and bind the lipoproteins to various cell membrane receptors or endothelial surface molecules, some are involved with activation or inactivation of various lipolytic enzymes such as lipoprotein lipase and other enzymes, and some serve as lipid acceptor proteins. Many of the apoproteins exist as genetically determined isoforms, which create individual and population differences in lipoprotein metabolism. Apart from apoproteins related to lipoprotein modulation, HDLs also transport numerous other proteins (over 50 have been identified), many of which are immunomodulatory in function. Collectively they are referred to as the HDL proteome [47].
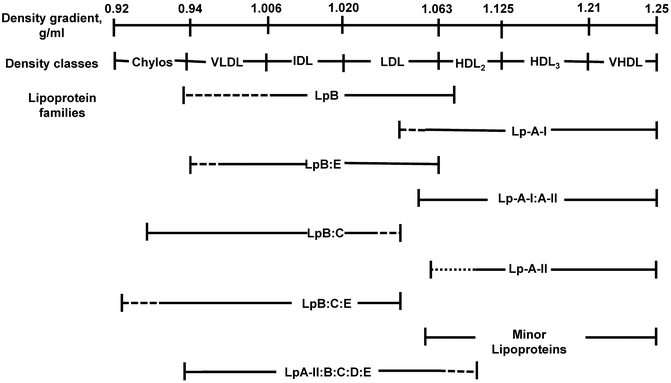
Fig. 4.6
The relationship of individual apolipoprotein A (apoA)- and apoB-containing lipoprotein (Lp) families defined by their unique apolipoprotein composition to major lipoprotein density classes against the density (d) gradient background (d = 0.92–1.25 g/mL). The lines under lipoprotein families designate the approximate density boundaries, with solid lines depicting the actual localization of each lipoprotein family and dotted lines the possible localization of each lipoprotein family. Lipoprotein families represent polydisperse systems of particles, each of which has a different lipid/protein ratio, but the same qualitative apolipoprotein composition. The polydisperse character of lipoprotein families is the main reason for their overlap within certain density segments (Chylos chylomicron, HDL high-density lipoprotein, IDL intermediate-density lipoprotein, LDL low-density lipoprotein, VHDL very high-density lipoprotein, VLDL very low density lipoprotein.) From Current Atherosclerosis Reports 2003, 5:459-467
Under normal physiologic conditions when lipoproteins have the proper core ratios of lipids, the following lipid trafficking pathways are operative. TG-rich chylomicrons are secreted into the lymphatic circulation (chylomicrons) where they make their way into plasma and join hepatically excreted TG-rich VLDLs. ApoA-V is part of TG-rich lipoprotein formation [48] and traffics with the particle as do multiple copies of apolipoprotein C-II (apoC-II), a ligand for LPL, and apolipoprotein C-I (apoC-I) and C-III (apoC-III) [49–51] (Fig. 4.7). ApoA-I is synthesized in hepatocytes and jejunal enterocytes and is secreted into plasma where it is rapidly lipidated but some apoA-I is also initially carried into plasma on chylomicron surfaces [52]. Delipidation of TG-rich particles or lipolysis occurs as TGs are hydrolyzed by lipoprotein lipase (LPL), a potent triglyceridase upregulated in large part by insulin in muscle and adipocyte vascular beds [53]. ApoA-V is important in the docking of TG-rich lipoproteins to heparan sulfate proteoglycans (HSPGs) in endothelial cell lipid rafts in the area of LPL expression and thus enhances lipolysis. ApoA-V is also involved with docking to the LDLr and LRP [54]. Chylomicron lipolysis is normally quite rapid due to the large size of these particles which contain multiple copies of apoC-II (an activator of LPL) resulting in smaller, TG-poorer particles called “chylomicron remnants.” Two proteins, lipase maturation factor 1 (LMF1) and glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1), regulate LPL maturation and binding and thus are important mediators of TG-rich lipoprotein lipolysis [55]. LPL is synthesized in the endoplasmic reticulum of myocytes and adipocytes where LMF 1 plays an essential role in the formation of catalytically active LPL, a process called lipase maturation, which then translocates to the luminal surface of endothelial cells where it binds to HSPGs. GPIHBP1 provides a platform for apoC-II binding to LPL. In vascular endothelial cells where GPIHBP1 is expressed, lipid rafts also express syndecan1 and fatty acid transporters such as CD-36 [56] (Fig. 4.8). ApoA-V also facilitates interactions between the TG-rich lipoproteins and GPIHBP1 [57].
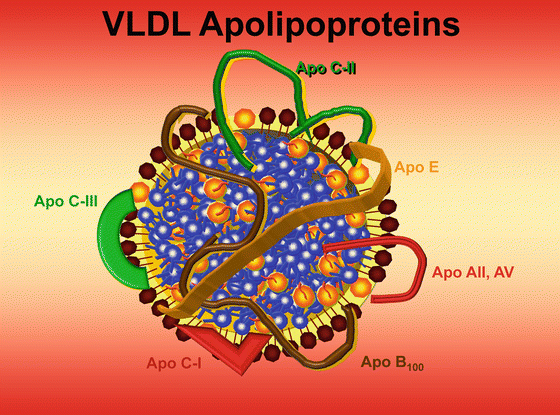
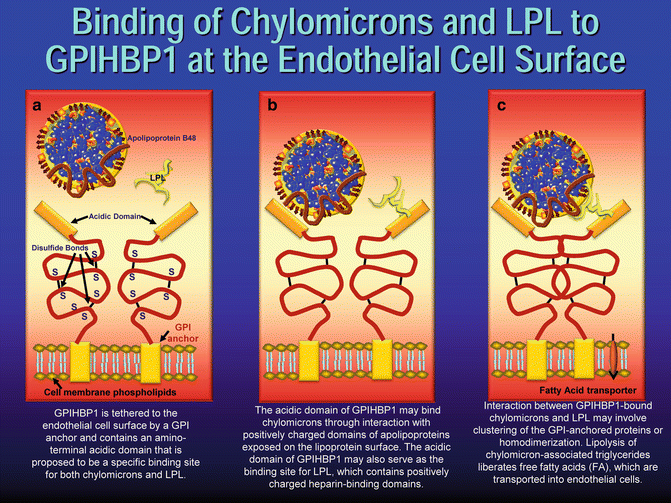
Fig. 4.7
Very-low-density lipoprotein is a large TG-rich particle that carries several apolipoproteins. C-II is a ligand for lipoprotein lipase. ApoC-I blocks LPL, HL, CETP, LCAT, VLDLr, and LDLr. ApoC-II binds to and activates LPL. ApoC-III interferes with apoC-II/LPL binding and apoE binding to receptors. ApoB is a ligand for LDL receptor. ApoE is a ligand for the LDL, VLDL receptor, and the LDL receptor-related protein (LRP). C-III inhibits the action of LPL and the ability of apoE to act with receptors. ApoA-II inhibits VLDL lipolysis and apoA-V helps bind TG-rich lipoproteins to HSPG in areas of LPL expression. Dayspring artwork
Fig. 4.8
Adapted from Ory DS. Chylomicrons and lipoprotein lipase at the endothelial surface (reference [56] in the chapter). Model for binding of chylomicrons and LpL to GPIHBP1 at the endothelial cell surface. (a) GPIHBP1 is tethered to the endothelial cell surface by a GPI anchor and contains an amino-terminal acidic domain that is proposed to be a specific binding site for both chylomicrons and LpL. (b) The acidic domain of GPIHBP1 may bind chylomicrons through interaction with positively charged domains of apolipoproteins exposed on the lipoprotein surface. The acidic domain of GPIHBP1 may also serve as the binding site for LpL, which contains positively charged heparin-binding domains. (c) Interaction between GPIHBP1-bound chylomicrons and LpL may involve clustering of the GPI-anchored proteins or homodimerization. Lipolysis of chylomicron-associated triglycerides liberates free fatty acids (FA), which are transported into endothelial cells
ApoA-IV contributes to lipolysis by facilitating efficient release of apoC-II from either HDL or VLDL, and once apoC-II is anchored by GPIHBP1, it binds to and activates LPL [53], resulting in hydrolysis of TG in chylomicrons or VLDL [58]. The resultant remnants are ultimately internalized by VLDL receptors in extrahepatic tissues [59] and by hepatic LDLr and LRP using apolipoprotein E (apoE) as a ligand. Another important regulator of TG-rich lipoprotein lipolysis is apoC-I which inhibits the binding of VLDL by LDLr, LRP or the VLDL receptor [60]. Such inhibition of TG-rich lipoprotein binding to lipoprotein receptors is thought to be due to the ability of apoC-I to alter or camouflage the conformation of apoE on TG-rich lipoprotein or to displace apoE from these particles. ApoC-I is also known to inhibit LPL, hepatic lipase (HL), phospholipase A2, as well as CETP [61], where it accounts for most of the CETP-inhibitory activity that is associated with human plasma HDL [62]. A chylomicron half-life is normally less than an hour, whereas VLDL lipolysis ranges from 2 to 6 h.
ApoC-III is synthesized in hepatocytes and enterocytes and is also a potent regulator of lipolysis [63]. It is present in three isoforms related to the number of sialic acid molecules (0, 1, or 2) terminating the oligosaccharidic portions of the protein, apoC-III0, apoC-III1, and apoC-III2. In plasma the isoform makeup is ~10, 55, and 35 % of the total apoC-III levels. ApoC-III1 and apoC-III2 correlate more with TG levels than apoC-III0 and apoC-III2 is associated with generation of small LDL. Collectively apoC-III stimulates VLDL assembly and secretion, inhibits LPL, in part by affecting binding of TG-rich lipoproteins to HSPG, and interferes with VLDLr, LRP, and LDLr uptake of lipoproteins [64, 65].
As the TG molecules are hydrolyzed by lipoprotein lipase to FA and monoacylglycerols, the TG-rich lipoproteins shrink, resulting in the loss of large amounts of their surface PL as well as some surface apolipoproteins. ApoC-III redistributes from VLDL to HDL and becomes ready for reuse and subsequent retransfer back to newly synthesized VLDL particles [66]. The now smaller particles carrying much less TG and PL still have their CE core: such particles are called VLDL and chylomicron remnants. The particles which were formerly TG-rich are now much less TG-rich and are trafficking primarily CE. The FA released from the TG can enter myocytes to be oxidized for energy, enter adipocytes and be reconverted to and stored as TG, or bind to albumin and be trafficked for use elsewhere. The PL can be taken up by the cell membranes or bind to phospholipid transfer proteins (PLTP) and carried to other cells or to maturing (lipidating) HDL particles. Ultimately, VLDL size decreases and density increases to the point where they become IDLs which under normal circumstances are rapidly removed by hepatic LDLr to which they dock via their apoB100 and apoE. This receptor attachment is aided by HL, which has both triglyceridase and phospholipase properties resulting in additional particle lipolysis creating apoB-containing LDL particles. LDL is a predominantly cholesterol-rich lipoprotein with a core TG/CE ratio of ≥4:1 [67]. The LDLs typically circulate for 1.5 to 3 days before most (90 %) are cleared by hepatic LDLr [68]. During their plasma residence time, LDLs are subject to homotypic exchange via CETP of their core CE for TG with VLDLs or heterotypic exchange with HDLs. CETP-mediated exchange of neutral lipids can be inhibited by apoC-I and apolipoprotein F (apoF) [69]. Since every cell in humans can synthesize cholesterol de novo, very little LDL-mediated delivery of cholesterol is necessary. Persons with hypobetalipoproteinemia have very low levels of LDL-C and suffer no cholesterol deficiency consequences [70]. Normally LDLs are cleared by LDLr binding to apoB and the process of LDLs returning their core CE to the liver, much of which originated in HDLs, is termed “indirect reverse cholesterol transport.”
The Apolipoprotein A-I Family of Lipoproteins
Aside from the apoB particles whose main mission is to traffic TG and phospholipids is the HDL family of apoA-I lipoproteins. Unlike chylomicrons and VLDLs, HDLs are created not in enterocytes or hepatocytes, but rather in plasma by the lipidation of secreted apoA-I and apolipoprotein A-II (apoA-II). Regulated primarily by peroxisomal proliferator-activated receptor-α (PPAR-α), apoA-I is produced and released by hepatocytes and enterocytes. The unique helical structure of apoA-I gives it an affinity for cholesterol binding. Lipidation occurs along cell membrane surfaces with excess UC; activation of the liver X receptor (LXR) in turn upregulates cell membrane cholesterol efflux proteins including ABCA1. Unlipidated apoA-I accepts effluxed UC and PL, creating pre-beta HDL species. ApoA-I lipidation activates lecithin–cholesteryl acyltransferase-α (LCAT-α) which catalyzes the transfer of fatty acids from the sn2 position of PL to the 3-hydroxy group on UC, changing the amphipathic UC into the hydrophobic CE. The molecular polarity change drives the CE away from the apoA-I particle surface of the HDL to its core explaining why, as the HDL matures, it converts from a discoidal to a spherical particle. ApoA-IV in HDL can activate LCAT and in free form in both lymph and plasma may also play critical roles in mediating ABCA1 cholesterol efflux. Additional HDL lipidation occurs via attachment of larger, more mature HDL species to ABCG1 sterol efflux transporters, SR-B1, or even by free diffusion from cells into larger HDL species [66]. As the HDL matures, it picks up, transfers, and reacquires numerous proteins (its proteome), including several apoproteins involved with lipoprotein catabolism and clearance including CETP, the apoC family, apoE, apolipoprotein A-II (apoA-II), apolipoprotein L (apoL), and apolipoprotein M (apoM), and others involved in a multitude of functions [71].
With respect to trafficking UC and CE, the vast majority (> 90 %) of HDL lipidation occurs via ABCA1 expression at the liver, small intestine, and adipocyte tissue [72].
In effect a serum HDL-C represents cholesterol derived from the gut and the liver and is not a reflection of peripheral cholesterol efflux. This suggests that HDLs evolved not solely to perform RCT but also delivery of hepatic- and enterocytic-derived UC elsewhere, primarily to steroidogenic tissues and adipocytes [73]. The likely reason HDLs have a 5-day half-life is to serve as a rapidly available supply of CE for the adrenal cortex under stressful hypercorticoid conditions like inflammation and infection [74] and as a repository for urgently needed immunoproteins. Because of those functions, many refer to HDLs as an innate part of the immune system [75]. A major part of HDL’s antiatherogenic potential is the ability to efflux both CE and UC from sterol-laden macrophages (foam cells) in atherosclerotic plaque referred to as macrophage reverse cholesterol transport (ΏRCT). Compared to total body cholesterol, the amount of cholesterol in plaque is very small, and ΏRCT, although cardioprotective, does not contribute significantly to a serum HDL-C value [76]. Other than trafficking cholesterol to the tissues mentioned above, HDLs through numerous pathways can facilitate fecal excretion of cholesterol. HDLs can return UC and CE to the liver where it is delipidated by SR-B1, or endocytosed by LDLr (using apoE as a ligand), or the ectopic β chain of ATP synthase (holoparticle) receptor [77]. HDLs can also be delipidated by a putative enterocyte receptor and the UC exported to the gut lumen via ABCG5 and ABCG8 transporters in a process now termed transintestinal cholesterol efflux or TICE [33]. HDLs returning cholesterol to the liver or gut is called direct RCT. However, a major part of RCT is HDLs heterotypically exchanging its CE for TG with apoB particles (chylomicrons, VLDLs, IDLs, and primarily LDLs). The apoB particles now carrying a CE load acquired from HDLs are cleared by the liver, in essence returning substantial cholesterol in what is now called indirect RCT. Total RCT is the sum of direct and indirect RCT and it should be clear that a serum HDL-C by itself has no relationship with this complex and dynamic HDL-mediated trafficking of cholesterol system [78]. The TG-rich HDLs undergo additional lipolysis utilizing HL and endothelial lipase [79]. During this process some apoA-I is shed and is cleared via cubilin and megalin in renal tubules [80]. In effect, apoA-I is constantly being synthesized, secreted, lipidated, delipidated, and ultimately cleared by the liver, gut, or kidney.
In summary, lipid homeostasis, fatty acids, and cholesterol, derived mostly from the liver and gut, but also peripheral cells, are trafficked as components of lipoproteins: FA for energy and cell membranes, and cholesterol for cell membranes and steroidogenesis. Excess cholesterol in the form of UC and CE is returned to the gut for fecal elimination or to the liver where UC is secreted into bile or converted to a bile acid for potential fecal excretion or become part of a newly formed VLDL or effluxed to a prebeta HDL. The system obviously is complex and mediated by dozens of genes, enzymes, proteins, and receptors, and pathology in any of those will negatively affect lipid (energy and sterol) homeostasis.
Insulin Resistance and Type 2 Diabetes
For the remainder of this chapter, lipoprotein pathophysiology related to insulin resistance (IR) and/or T2DM will be reviewed. A normal person is sensitive to the hormone insulin which regulates carbohydrate and fatty acid metabolism, lipogenesis, lipolysis, and, hence, energy homeostasis. Insulin mediates the uptake of glucose into cells where in muscles and liver it can be converted to and stored as glycogen. In IR there is impaired signaling via the phosphoinositol-3 kinase pathway allowing the buildup of toxic lipid metabolites, such as FA acyl CoA, diacylglycerol, and ceramide in numerous tissues including the liver, pancreatic beta cells, and adipocytes [81]. It is the IR-related lipid-mediated macrovascular complications, in large part related to atherothrombotic events, that result in the high morbidity and mortality risk seen in T2DM.
Cholesterol Synthesis and Absorption: Major epidemiological trials like Framingham Offspring [82], PROCAM [please define acronym 83], and Cardiovascular Risk in Young Finns Study [84] have related increased CV risk in patients with increased levels of phytosterols which are measurable markers of sterol absorption. Miettinen showed that in the Scandinavian Simvastatin Survival Study (4S), a high-risk secondary prevention trial of statin-/placebo-treated patients with high LDL-C, hypoabsorbers did and hyperabsorbers did not have a beneficial effect of simvastatin, theoretically because hyperabsorbers are typically hyposynthesizers of cholesterol and therefore less likely to be responsive to a statin [85]. In the DEBATE [Drugs and Evidence-Based Medicine in the Elderly] study, low-cholesterol absorption was associated with fewer recurrent cardiovascular events and with better survival in elderly patients despite frequent abnormalities of glucose metabolism [86]. Intestinal function is abnormal in diabetics and several enterocytic sterol homeostatic regulatory changes occur in IR patients. Conflicting studies have described T2DM as having either reduced [87] or increased cholesterol absorption [88]. A recent study demonstrated cholesterol absorption was highest in the lean insulin-sensitive participants, whereas cholesterol synthesis was highest in the lean IR and obese IR participants [89]. In another experiment 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) reductase, the rate-limiting enzyme for cholesterol synthesis, is increased in animal models of diabetes in both the liver and small intestine [90].
Rats made diabetic by injection of streptozotocin are hyperabsorbers of cholesterol which was explained by changes in intestinal absorption-regulating proteins, namely, an upregulation of NPC1L1, ACAT2, and microsomal triglyceride transfer protein (MTP) and a reduced expression of ABCG5 and ABCG8 [91]. Lally et al. showed that diabetic patients had more NPC1L1 mRNA than the control subjects (p < 0.02) and expression of ABCG5 and ABCG8 mRNA was lower in the diabetic patients (p < 0.05) and MTP expression was increased (p < 0.05). There was a positive correlation between NPLC1L1 and MTP mRNA (p < 0.01) and a negative correlation between NPC1L1 and ABCG5 mRNA (p < 0.001) [92]. In addition, an increase in apolipoprotein B48 synthesis has been demonstrated in animal models of diabetes and insulin resistance. Generally, apoB synthesis and utilization is driven by increased lipid substrate availability. Hyperabsorption is a manifestation of intestinal dysfunction and leads to abnormal chylomicron composition which, via the action of CETP, will directly influence the composition of other circulating lipoproteins [93].
Experts have speculated on whether knowing one is or is not a hyperabsorber or hypersynthesizer of cholesterol would be useful in deciding on lifestyle and drug therapies, and there are both null and supporting data on this. Certainly, statins and statins plus cholesterol absorption inhibitors such as ezetimibe improve lipid and lipoprotein abnormalities in T2DM [94]. Of interest is that potent statin monotherapy can significantly increase cholesterol and noncholesterol sterol absorption, which has the potential to offset some of the benefit of inhibiting synthesis [95].
The TG/HDL Axis: ApoB-Containing Lipoproteins
The major lipid abnormality in T2DM has been called a TG/HDL axis disorder and is characterized by variable TC and LDL-C but elevations of fasting and often postprandial TG and reductions in HDL-C [96]. Underlying these lipid concentration abnormalities are significant changes in the number, size, core lipid composition, and proteome of lipoproteins. It then becomes crucial to understand what happens to previously described lipoprotein genesis and trafficking of lipids in the IR patient with TG/HDL abnormalities. Clinicians are going to have to respect the pathology related to TG-rich lipoproteins and TG levels which heretofore were not deemed to be of concern (<150 mg/dL). Typically the liver in IR patients has increased pools of retained lipids, especially that of TG which results from an imbalance between the uptake and synthesis of fatty acids and their oxidation and export [97]. Both hyperinsulinemia and hyperglycemia induce the expression of the lipogenic gene-activating hepatic sterol regulatory element-binding protein 1c (SREBP1c) and the carbohydrate-responsive element-binding protein (ChREBP) [98].
The more lipid substrate (especially triglycerides) available in the hepatocyte or enterocyte cytosol, the more apoB will be lipidated rather than catabolized (Fig. 4.9). Insulin reduces MTP expression via activation of the mitogen-activated protein kinase (MAPK) pathway [99]. Normally, lipidation of apoB creates a primordial VLDL that evolves into a normally composed, sized, and secretable VLDL2 [100]. Secretion of VLDL2 is the same in IR and insulin-sensitive subjects. When there is a lack of lipid pool, there is improper folding and rapid degradation of apoB and less VLDL is produced [101]. The major cause of hypertriglyceridemia in the HOMA-IR person is the increased availability of free FA substrate causing hepatic overproduction and secretion of larger TG-rich VLDL1, resulting in increased plasma concentrations of apoB and TG [102] (Figs. 4.9 and 4.10). A recent nutritional study demonstrated apoB production had a strong relation with dietary fructose and especially fructose corn syrup and not glucose [103]. In a kinetic study, plasma glucose, insulin, and free fatty acids together explained 55 % of the variation in VLDL1 TG production rate [104]. The large VLDL1 seen in T2DM are normally suppressed by insulin, but not when IR is present. The apoB100-containing VLDL2 are converted to VLDL1 by the addition of a major load of triglycerides in the endoplasmic reticulum (the same is true of enterocyte apoB48 and chylomicron formation). VLDL1 creation also is dependent upon ADP ribosylation factor 1 (a small GTP-binding protein) which is involved with translocation from ER to the Golgi apparatus where final synthesis including much of the TG acquisition and phospholipidation occurs [105, 106]. The time between apoB100 production and lipidation to create large VLDL1 is approximately 15 min [105]. Insulin resistance results in diminished phosphatidylinositol-3-kinase that may add to the increased VLDL secretion [107]. In humans, the mean residence time of VLDL1 apoB is the main determinant of apoB pool size and of plasma TG concentration [108]. There is an increased production of VLDL1, as well as a reduction in the catabolic rate of apoB-containing lipoproteins, in particular IDL and LDL. Collectively this leads to increased levels of apoB related to large VLDL-P and LDL-P. The catabolism of apoA-I, the main apolipoprotein of HDL, is increased by 48 %, but apoA-I production is increased by 25 %, probably because of some compensatory effect. This production/clearance imbalance results in a 16 % reduction in the concentration of HDL apoA-I [109] (Fig. 4.11). Garvey et al. in an elegant insulin clamp study analyzing NMR-derived particle concentrations showed that as the patients’ status progressed from insulin sensitive to insulin resistance to T2DM, there are increases of VLDL-P, IDL-P, and most especially LDL-P [110].
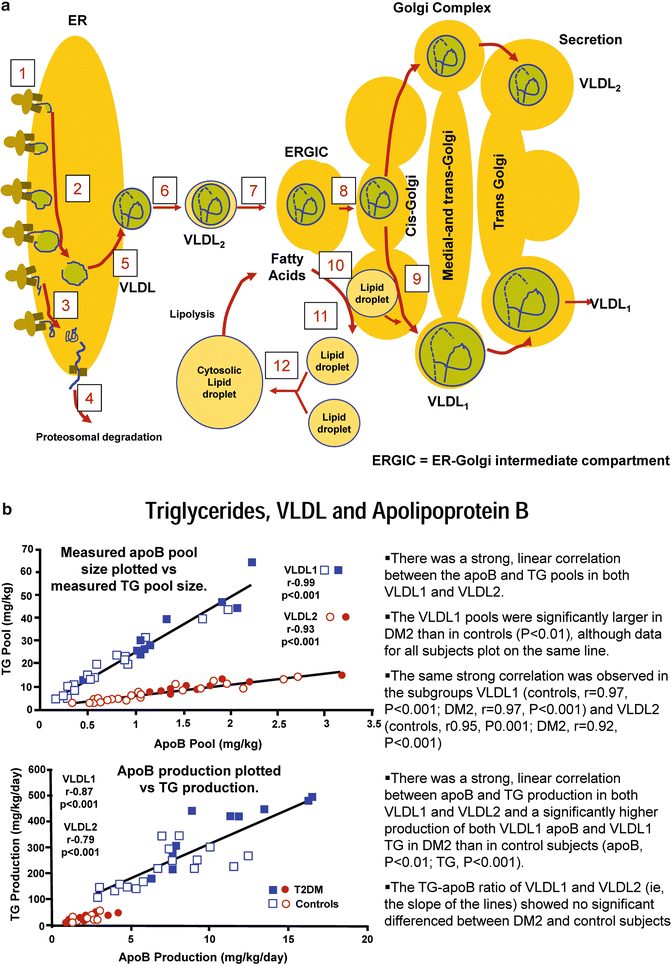
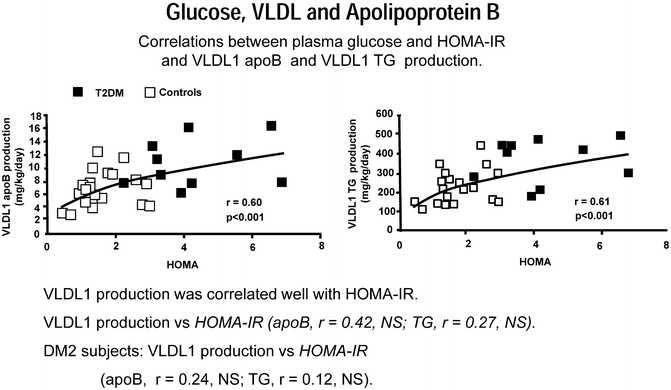
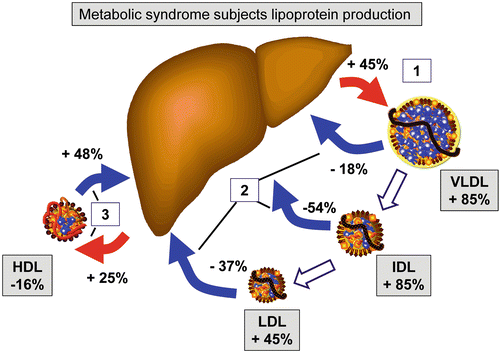
Fig. 4.9
(a) Adapted from Arterioscler Thromb Vasc Biol. 2008;28:1225-1236 [reference 109 in chapter]. The assembly process starts in the rough endoplasmic reticulum (ER) by the biosynthesis and concomitant (cotranslational) translocation of apolipoprotein B100 (apoB100) to the lumen of this organelle. ApoB100 interacts cotranslationally with the microsomal triglyceride transfer protein (MTP) and is thereby lipidated to form a primordial particle (pre-VLDL). Alternatively, apoB100 fails to be lipidated and misfolds. This results in a sorting to degradation. Thus, the protein is unfolded and retracted to cytosol, ubiquitinated, and sorted to proteasomal degradation. Pre-VLDL is converted to VLDL2 late in the ER compartment. VLDL2 exits the ER at specific exit sites of this organelle by Sar1/Cop II vesicles, which fuse to become the ER–Golgi intermediate compartment (ERGIC) (7). ERGIC fuses with Cis-Golgi. In the Golgi apparatus, VLDL2 is converted to VLDL1 by the addition of a bulk load of triglycerides. This lipidation process differs from that which gives rise to pre-VLDL and VLDL2. The formation of VLDL1 may involve the formation of a lipid droplet in the lumen of the secretory pathway. The mechanism behind the formation of lipid droplets in the secretory pathway may follow that of cytosolic lipid droplets. Such droplets are formed from the microsomal membranes under the influence of the enzymes phospholipase D1 and ERK2 as well as of adipocyte differentiation-related protein (also known as adipophilin and caveolin). The formation of the cytosolic droplets also involves a fusion step that is dependent on microtubules and the motor protein dynein. (b) Triglycerides, VLDL, and apolipoprotein B. Adapted from Adiels M et al. Arterioscler Thromb Vasc Biol. 2005;25:1697-1703 [Reference 104 in chapter]
Fig. 4.10
Glucose, VLDL, and apolipoprotein B. Adapted from Adiels M et al. Arterioscler Thromb Vasc Biol. 2005;25:1697-1703 [Reference 104 in chapter]
Fig. 4.11
Adapted from Arterioscler Thromb Vasc Biol. 2008;28:1225-1236 [reference 109 in chapter]. Changes in lipoprotein metabolism in T2DM and the metabolic syndrome. Subjects diagnosed with the metabolic syndrome display, most noticeably, an increased production of VLDL (1), and there is a reduction in the catabolic rate of apoB-containing lipoproteins, in particular IDL and LDL (2). Together, these result in increased concentrations of apoB-containing lipoproteins. The catabolism of apoA-I, the main apolipoprotein of HDL, is increased by 48 %, but apoA-I production is increased by 25 %, probably because of some compensatory effect (3). This results in a 16 % reduction in the concentration of HDL apoA-I
VLDL lipolysis is delayed in T2DM due to several mechanisms. As previously mentioned several apolipoproteins are involved with efficient VLDL catabolism including apoE, apoA-II, apoA-IV, apoA-V, ApoC-I, apoC-II, apoC-III, apoD, and apoF. Many of those apolipoproteins have altered function in IR persons and T2DM. Ultimately lipolysis of TG-rich lipoproteins requires apoC-II to activate LPL and thus release of apoC-II from either HDL or VLDL allows for LPL-mediated hydrolysis of TG in nascent chylomicrons and VLDLs [111]. In a small study of diabetic patients vs. normolipemic controls who had TG-tolerance tests, the diabetics displayed typical postprandial hypertriglyceridemia, but although apoA-V levels were similar in the two groups, paradoxically the diabetics had increased postprandial apoA-V in non-HDL particles which is suggestive that apoA-V is not involved in the regulation of TG in the postprandial state [112]. In another study, the postprandial (after an oral fat load) increase of apoA-V was confirmed and was related to plasma TG and VLDL1-TG but also to apoC-III. It was thought the increase of apoA-V simply reflected the increase of VLDL particles related to apoC-III overproduction [113]. ApoA -V genotypes do not appear to have an impact on risk of development of T2DM [114].
ApoC family members are crucial to the synthesis of TG-rich lipoproteins as well as their lipolysis and catabolism. High concentrations of ApoC-I and ApoC-III are associated with increased triglycerides in men with the metabolic syndrome. These findings in humans were first seen in Hyplip2 congenic mouse strain studies which related the elevated TG to delayed catabolism of VLDL, which in turn led to decreased FA delivery to visceral adipose tissue [115]. In obese males with the metabolic syndrome, apoC-I and ApoC-III levels were mainly related to the visceral adipose tissue (VAT) compartment (measured using nuclear magnetic resonance). This was related to a higher expression of LPL in VAT versus subcutaneous adipose tissue (SAT). The ApoC-I and ApoC-III inhibition of LPL therefore contributed to both higher TG and lower VAT area in human subjects. The difference in effect of ApoC-I and ApoC-III on TG concentrations in this study underlines the stronger inhibition of LPL by ApoC-III compared with ApoC-I [116] (Figs. 4.12 and 4.13).
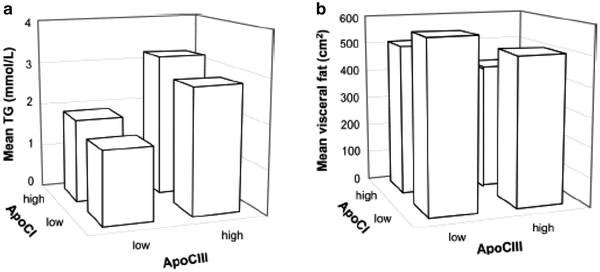
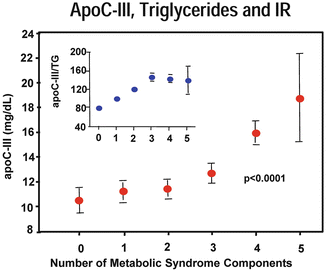
Fig. 4.12
Illustration of the inverse relationship of ApoC-I and ApoC-III concentrations with plasma triglyceride concentration (positive, a) and with visceral adipose tissue area (negative, b). ApoC-I/ApoC-III median/mean low, ApoC-I/ApoC-III median/mean high. Groups: (1) low ApoC-I and ApoC-III (n = 35), (2) high ApoC-I/low ApoC-III (n = 21), (3) low ApoC-I/high ApoC-III (n = 14), and (4) high ApoC-I and ApoC-III (n = 28). Triglyceride concentration: 1 vs. 3, 1 vs. 4, and 2 vs. 4: p < 0.001. 2 vs. 3: p < 0.01. Visceral adipose tissue area: low ApoC-I and apoC-III vs. high ApoC-I and ApoC-III: p < 0.001. From van der Ham RL, Alizadeh Dehnavi R, Berbée JF, Putter H, de Roos A, Romijn JA, Rensen PC, Tamsma JT. Diabetes Care. 2009;32:184-6 [Reference 116 in chapter]
Fig. 4.13
Mean of apoC-III concentration according to the number of metabolic syndrome components. Data normalized for TG values (figure inset) show statistically significant trends for the apoC-III/TG ratio (p < 0.0001). Hermes Florez et al. Atherosclerosis 2006;188:134-141
All apoCs are distributed in a cycling process between TG-rich apoB lipoproteins and HDL. In the fasting state, apoC-II and apoC-III are equally distributed between HDL and VLDL, whereas apoC-I is mostly trafficked with HDL (>90 %). Thus, in the exchange of apolipoproteins after a meal, more apoC-I is transferred than other apoCs. The apoC-I enrichment of TRLs after a meal affects particle catabolism and is involved with the formation of VLDL and chylomicron CE-rich atherogenic remnants. A paradox is that apoC-I is not known to interfere with TG hydrolysis as studies have shown that apoC-I-enriched TRLs undergo normal hydrolysis forming smaller TRLs and remnants. Because apoC-I is a potent inhibitor of apoE-mediated uptake of TG-rich lipoproteins by LDLr, VLDLr, and LRP, particle clearance is impaired. ApoC-I-enriched particles which have compositional abnormalities (TG-rich) have increased plasma residence time allowing CETP-mediated exchange of core TG for CE utilizing heterotypic and homotypic pathways which over time make the remnants even more CE-rich. Numerous studies have demonstrated that CE-rich remnants are atherogenic, and delayed remnant clearance during the postprandial state is a well-established feature of patients with coronary artery disease (CAD) [117]. Interestingly ApoC-I is a more potent inhibitor of CETP when it is on HDL but not the apoB particles. Thus, the transfer of apoC-I from HDL to TG-rich lipoproteins facilitate atherogenic remnant formation, suggesting a dual role of apoC-I: (1) preventing remnant formation and premature atherosclerosis if attached to HDL and (2) promoting remnant formation and atherosclerosis if transferred to TG-rich lipoproteins [118]. ApoC-I content of postprandial TG-rich lipoproteins has been shown to be a risk factor for early atherosclerosis in normolipidemic healthy middle-aged men, supporting the conclusion that the enrichment of remnant lipoproteins with cholesterol is not favorable. ApoC-I on TG-rich lipoproteins has been linked to increased CIMT [119] (Fig. 4.14). There are not a lot of published studies evaluating apoC-I, per se, in diabetes. In an evaluation of women with PCOS, those with IR were characterized by statistically significantly elevated levels of apoC-I compared with those of non-IR patients. ApoC-I correlated with BMI, TG, HDL-C, apoA-I, and HOMA-IR [120].
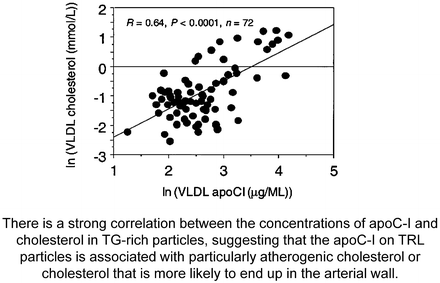
Fig. 4.14
From Hamsten A, Silveira A, Boquist S, Tang R, Bond G, de Faire U, Björkegren J. The apolipoprotein CI content of triglyceride-rich lipoproteins independently predicts early atherosclerosis in healthy middle-aged men. J Am Coll Cardiol 2005;45:1013-7 [Reference 119 in chapter]
ApoC-III is perhaps the most complex and enigmatic apolipoprotein. For some time, it has been known that apoC-III-enriched particles were a significant CHD risk factor. ApoC-III levels are associated with hypertriglyceridemia, increases in VLDL-P and VLDL-TG, and inversely related to the size of LDL particles [121]. In the Cholesterol and Recurrent Events (CARE) trial, both the plasma concentrations of VLDL particles and apoC-III in VLDL and LDL were better predictors of coronary heart disease risk than was plasma TG [122] (Fig. 4.15). In CARE diabetic status compared to nondiabetic status per se was not associated with high concentrations of apoC-III-containing TG-rich lipoprotein particles, if their plasma TG levels were similar [123]. Because the apoC-III location on chromosome 11 is near insulin response elements, a link to diabetes has been surmised [124]. Several nuclear transcription factors (NTF) influence apoC-III. One is Foxo1 which provides a molecular link between insulin resistance and the pathogenesis of diabetic hypertriglyceridemia. Foxo1 is a substrate of Akt/protein kinase B and glucocorticoid inducible kinase, which is involved with insulin signaling and in modulating both hepatic and intestinal apoC-III expression. Under both insulin-deficient and insulin-resistant conditions, Foxo1 expression is deregulated, contributing to the increased apoC-III production and impaired plasma TG metabolism [125]. Hepatic nuclear factor 4-alpha (HNF-4α) which regulates LPL is also a strong positive regulator of apoC-III expression [126]. HNF-4α is stimulated by glucose and the carbohydrate-responsive element-binding protein (ChREBP). In individuals with IR and diabetes, there is a loss of insulin-mediated suppression of apoC-III that, coupled with glucose-stimulated apoC-III expression, leads to hypertriglyceridemia [127, 128] (Fig. 4.16). New findings demonstrate that apoC-III can play an additional “feedback” role in PPAR-α -mediated metabolic and inflammatory functions by controlling lipolytic generation of PPAR-α ligands. Because apoC-III expression is suppressed and LPL activity is stimulated by PPAR-α, a positive feedback system may exist. Individuals with high apoC-III levels may have impaired generation of endogenous PPAR-α ligands. Such a scenario is likely is patients with IR [129].
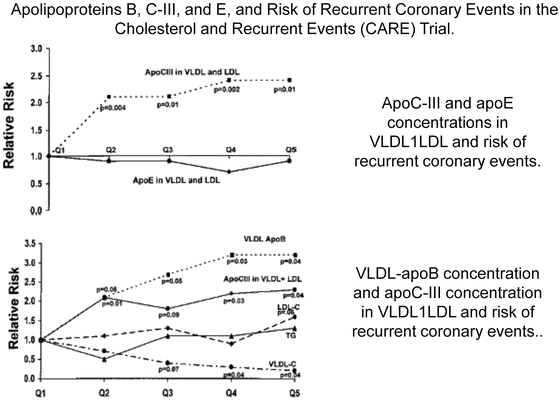
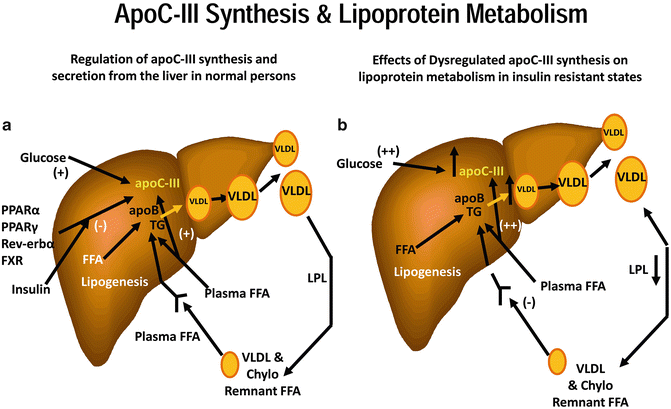
Fig. 4.15
Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, Pfeffer MA, Braunwald E. VLDL, Circulation. 2000;102:1886-1892 [Reference 122 in chapter]
Fig. 4.16
 Laboratory Assessment of Lipoproteins in Diabetes
Lipoprotein Subclasses and Cardiovascular Disease Risk in Insulin-Resistant Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Lipoprotein (LDL and HDL) Oxidation in Diabetes Mellitus
Lipoproteins and Diabetic Nephropathy
Laboratory Assessment of Lipoproteins in Diabetes
Lipoprotein Subclasses and Cardiovascular Disease Risk in Insulin-Resistant Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Lipoprotein (LDL and HDL) Oxidation in Diabetes Mellitus
Lipoproteins and Diabetic Nephropathy




From Ginsburg and Brown. Arterioscler Thromb Vasc Biol 2011;31;471-473 [Reference 127 in chapter]. (a) Under normal conditions, apoC-III gene expression and synthesis are regulated by several factors, including nuclear transcription factors PPAR-α, PPARγ, Rev-erb, farnesoid X receptor, as well as insulin and glucose. All are inhibitory except for glucose, which stimulates apoC-III expression. Plasma free fatty acids (FFA) stimulate apoC-III secretion, but it is not known whether this occurs at the transcriptional or posttranslational level. ApoC-III in plasma inhibits lipoprotein lipase-mediated catabolism of VLDL (and chylomicrons) and inhibits the uptake of VLDL (and chylomicron) remnants by the liver. In addition, apoC-III may increase VLDL assembly and secretion. (b) In states of insulin resistance, any inhibitory role of insulin on apoC-III expression may be lost, whereas higher glucose levels, particularly in patients T2DM, would further stimulate apoC-III expression. Increased plasma FFA delivery to the liver would exacerbate this problem. The results of dysregulated apoC-III synthesis and secretion would be defective LPL-mediated lipolysis of TG-rich lipoproteins and reduced remnant lipoprotein clearance. Thus, dysregulated apoC-III synthesis and secretion could play a major role in the genesis of the diabetic, insulin-resistant dyslipidemia. In addition, accumulation of apoC-III-rich apoB-containing lipoproteins might have direct atherogenic consequences
Related posts:
Laboratory Assessment of Lipoproteins in Diabetes
Lipoprotein Subclasses and Cardiovascular Disease Risk in Insulin-Resistant Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Lipoprotein (LDL and HDL) Oxidation in Diabetes Mellitus
Lipoproteins and Diabetic Nephropathy
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
