Fig. 5.1
The lipoprotein cascade
Apolipoproteins and Triglyceride-Rich Lipoprotein Metabolism
So far we have given a very simplified version of lipoprotein metabolism. Apolipoproteins other than apo B48 and apo B100 are important for the chylomicron and VLDL story. Apo E, for example, which is synthesised in the liver and other extrahepatic tissues including macrophages, is transferred to the chylomicron and VLDL particles in the circulation. Apo E is necessary for clearance of the triglyceride-rich particles by the B/E receptor in the liver. Chylomicrons from diabetic patients have less apo E per particle than those from control subjects [6]. Once released from the chylomicron particle, or indeed from the VLDL particle since apo E is also attached to VLDL and is involved in its uptake by the liver, apo E is transferred to HDL. Apo E is a very interesting protein in that it appears also to mediate cellular cholesterol efflux when attached to apo B [7]. (Many extrahepatic cells including the macrophage secrete apo E) [8].
Apo E recycling in the hepatocyte is associated with an increase in ABCA1, a mechanism by which apo E increases cholesterol uptake by the liver [7]. Apo CI is another apolipoprotein attached to the chylomicron and VLDL; 70 % of apo CI is associated with HDL. During the postprandial rise of triglyceride-rich lipoproteins in serum, apo CI is transferred from HDL to VLDL [9]. Apo CI, at least experimentally, modulates lipoprotein production by increasing the production rate of hepatic VLDL, inhibiting lipoprotein lipase activity, interfering with apo E-mediated uptake of VLDL and inhibiting cholesteryl ester transfer protein (CETP). CETP transfers cholesterol from HDL to VLDL in exchange for triglyceride (for review see Tall [10]). Apo CII on the other hand is a cofactor for lipoprotein lipase which hydrolyses the triglyceride in chylomicrons and VLDL and promotes their uptake by the liver receptors and thus is associated with a decrease in triglyceride-rich lipoproteins. ApoC-III is yet another constituent of triglyceride-rich lipoproteins which impairs lipoprotein uptake and is involved in hypertriglyceridemia and fatty liver disease [11]. It has also been shown to enhance hepatic triglyceride-rich VLDL assembly and secretion under lipid-rich conditions [12].
Low-Density Lipoprotein
The LDL particle is a cholesterol-rich, triglyceride-poor particle (Fig. 5.2). LDL is composed of a hydrophilic surface layer of phospholipid, free cholesterol and hepatically derived apo B100 which packages the particle and adds stability. The core of the LDL particle includes esterified cholesterol and triglyceride together with the fatty acid tails of the phospholipid. LDL may act as a carrier for other insoluble particles such as free fatty acids and proteins which may be loosely attached [13]. Perhaps more importantly, lipoprotein lipase attaches to the particle and facilitates attachment of the particle onto the endothelial cell surface.
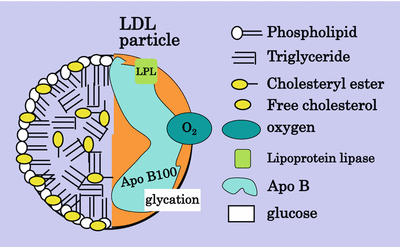
Fig. 5.2
The LDL particle
LDL can be subdivided into sizes by gradient gel electrophoresis and separated into a pattern A and a pattern B, pattern B being termed small dense LDL [14]. This pattern has been associated with an increase in atherosclerosis, but it has been difficult to define changes in composition of the LDL that create the increased atherogenicity. The usual way to separate the different sizes of apo B-containing lipoproteins is by ultracentrifugation, but the correlation between the denser particles on ultracentrifugation and electrophoresis is uncertain. The most recent addition to the methods to investigate lipoproteins is magnetic resonance (MR) spectroscopy which can sort particle size in large numbers of samples over very short time, but this technique still does not define small dense LDL [15]. Some years ago a subfraction of LDL with oxidised characteristics was described and was named electronegative LDL (LDL−) based on its properties of electrical mobility [16]. It was later renamed minimally oxidised LDL. More heavily oxidised LDL is more electronegative than LDL− and is identified as LDL− −. It now appears that electronegative LDL may also be produced by phospholipase (PL) A2. Rosenson et al. in the PLASMA11 Trial [17] showed that an inhibitor of PLA2 reduced LDL by 7 % and small dense LDL by 11 %. Enrichment of LDL with apoC-III contributes to the electronegativity [18]. Anti-LDL-monoclonal antibody had a protective effect against atherosclerosis in LDL receptor knockout mice [19]. It has been suggested that LDL− is a potential stress biomarker present in health and disease [20]. Small dense LDL isolation by various methods has been compared by Cheung [21]. The suggestion is that LDL’s atherogenicity resides in the large amount of cholesterol being packaged in a relatively small volume; hence, the surface area of the particle is relatively large making it more easily amenable to modification and therefore more avidly taken up by scavenger receptors. Small dense LDL is also more susceptible to glycation even in non-diabetic people [22]. The association between small dense LDL and VLDL has been investigated, not least because of the difficulty of demonstrating hypertriglyceridemia as an independent risk factor for atherosclerosis. VLDL, like LDL, comes in many sizes depending on its triglyceride load. The Scottish and Finnish groups [23, 24] many years ago demonstrated the relationship between large triglyceride-rich VLDL and small dense LDL: the larger the VLDL, the smaller and denser the LDL. Oxidation of the LDL particle depends on oxidation of its constituent protein and/or fatty acids. Polyunsaturated but not monounsaturated fatty acids are amenable to oxidation; hence, a particle rich in linoleic acid is more susceptible to oxidation than one rich in oleic acid [25] (Fig. 5.3).
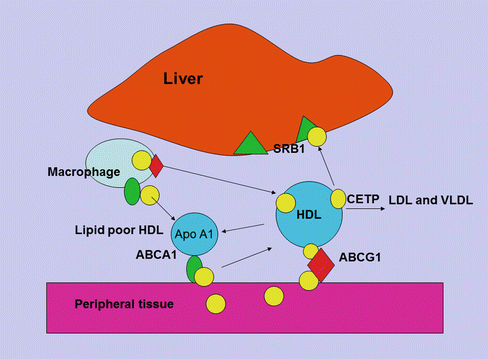
Fig. 5.3
HDL cholesterol uptake by the liver. Cholesterol uptake by HDL from macrophages and peripheral tissue is facilitated by ABCA1 and ABCG1 receptors. The HDL particle then docks with the liver giving up free cholesterol and lipid through the scavenger receptor (SR) B1 pathway and becomes free to circulate as nascent HDL
High-Density Lipoprotein
Apoprotein AI is the major apoprotein in HDL and functions as a cholesterol acceptor in the periphery through a complex set of interactions. ATP-binding cassette transporter A1 (ABCA1) facilitates the efflux of cellular phospholipid and free cholesterol to assemble with apolipoprotein AI (apo A-I), forming nascent HDL particles. ABCG1 is another protein involved in cholesterol efflux from peripheral tissue to apo A-I for reverse transport and binds larger, more spherical HDL species. Lipid-poor apo A-I accepts cholesterol released from macrophages forming nascent HDL [26]. The esterification of cholesterol to cholesteryl esters by lecithin/cholesterol acyltransferase (LCAT) is important for the process of mobilizing cholesterol from the periphery. And once HDL becomes mature, it may transfer cholesterol and phospholipid, through the action of cholesteryl ester transfer protein (CETP) and phospholipid transfer protein (PLTP) to apo B-containing lipoproteins, in exchange for triglyceride which is then hydrolyzed by the action of hepatic lipase [27]. The HDL particle docks with the liver and gives up its remaining cholesterol and lipid through the scavenger receptor (SR) B1 pathway and becomes free to circulate as nascent HDL. There is an inverse relationship between HDL cholesterol and hepatic expression of SR-B1 [28]. In passing it should be noted that the kidney plays an important part in apo A-I metabolism by both synthesis and clearance [29]. Apo AII is another apoprotein constituent of HDL and facilitates cholesterol efflux, HDL remodelling and cholesteryl ester uptake. Apo AII has been found to be a strong risk factor for cardiovascular disease, and it has been suggested that variation in Apo AII production may exert an influence on apo B production [30]. The composition of HDL reflects on its various functions. For example, its ability to act as an antioxidant to apo B-containing lipoproteins through PON-I [31] and reconstituted HDL has been shown to have an anti-thrombotic effect [32]. HDL may play a role in inflammation, and recently it has been shown that serum amyloid A, which is elevated in inflammation and may be deposited in atheroma plaque, may promote endothelial dysfunction. HDL may reverse this process at least to some extent [33]. More recent studies have suggested that HDL may modulate glucose metabolism in the muscle and affect insulin secretion [34].
Diabetes, Insulin Resistance and the Metabolic Syndrome
The Chylomicron
To explore the effect of diabetes and insulin resistance on lipoproteins, we will start at the beginning. In this chapter the beginning must be the chylomicron and its synthesis, since without food there would be little interest in diabetes or insulin resistance and in the rural areas of the world where starvation occurs, there is little talk of type 2 diabetes or insulin resistance. The apo B48-containing chylomicron transports both cholesterol and triglyceride from the intestine to the circulation and has a dominant role in distributing fatty acids/triglyceride to the tissues prior to being taken up by the liver [35]. The second function of the chylomicron is to transport cholesterol to the liver, although on the way the cholesterol may be taken up by tissues including the atheromatous plaque where the macrophage sits in waiting with a specific apo B48 receptor [36] as well as VLDL and scavenger receptors [35]. Apo B48 has been demonstrated in plaque by a number of workers [37–40]. Apo B48 is the solubilising protein necessary for the transport of cholesterol and lipid in aqueous solution in humans. The amount of triglyceride available for the chylomicron particle is limitless, the normal gut managing to limit the amount of triglyceride/fatty acids in the stool to under 5 g/day. On the other hand serum cholesterol is very tightly regulated and varies very little throughout one’s lifetime due to a hugely efficient regulatory process. Absorbed cholesterol varies considerably from person to person, and high absorbers of cholesterol have been shown to have low synthesis rates and to be less sensitive to cholesterol lowering with statin therapy [41]. The mechanism that regulates cholesterol absorption in the intestine is complex and both diabetes and insulin resistance have been shown to affect the regulation [42, 43], causing the initiation of the dyslipidemia of insulin resistance and diabetes (Fig. 5.4).
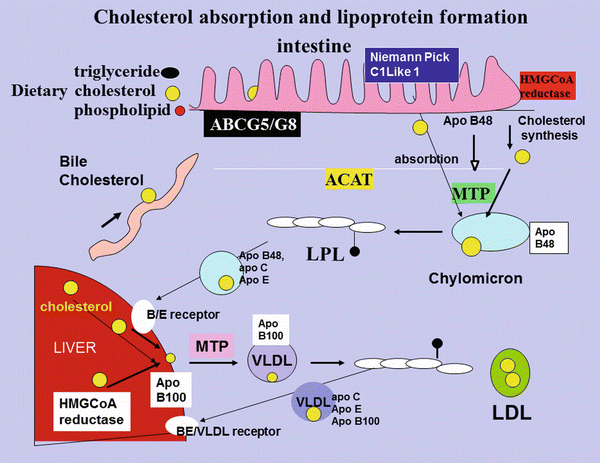
Fig. 5.4
Cholesterol absorption and lipoprotein formation. Dietary cholesterol, biliary cholesterol and cholesterol synthesized in the intestine for which HMGCoA is the rate-limiting enzyme, is transported across the cell membrane by NCP1-L1 and, together with triglyceride, phospholipid and the intestinally derived apoB48 protein, is assembled, under the influence of MTP into the triglyceride-rich chylomicron. Some of the absorbed cholesterol is excreted back into the lumen of the intestine under the influence of ABC G5/G8. The chylomicron is partially hydrolyzed in the circulation by lipoprotein lipase and acquires apoC-III and apo E. The resulting chylomicron remnant is taken up by the B/E receptor in the liver. The cholesterol and triglyceride released are reassembled with hepatically synthesized cholesterol and apo B100 to form VLDL. Lipoprotein lipase in the artery wall releases the triglyceride from VLDL and it acquires apoC-III and apo E. Some of the VLDL is taken up again by the liver, and the rest is further hydrolysed and looses apoC-III and E to become IDL and then LDL
Intestinal Niemann-Pick C1-Like 1 Protein
The first step in cholesterol absorption in the intestine appears to be through the multi-transmembrane protein Niemann-pick C1-like 1 (NCP1-L1) which is highly expressed in the jejunum. In humans it is localised to the brush borders of the enterocytes and acts as a unidirectional transporter of cholesterol and non-cholesterol sterols [44, 45]. The mechanism of action of NCP1-L1 has been elucidated. It has been shown that cholesterol promotes the formation and endocytosis of NCP1-L1-flotillin-cholesterol membrane microdomains which is an early step in cholesterol uptake. Zang et al. [46] discovered that it is the N-terminal domain of NCP1-L1 that binds cholesterol. It is interesting that this domain does not bind to plant sterols; thus, it now seems that plasma membrane-bound NCP1-L1 binds exogenous cholesterol, and this binding facilitates the formation of the NCP1-L1-flotillin-cholesterol microdomains that are then internalised into cells through the clathrin adaptor protein 2 pathway. Twenty rare NCP1L1 alleles have been found in the low cholesterol absorbers and appear to impair NCP1-L1 cholesterol uptake through various mechanism ([47, 48]; for review see Calandera [49]). It has been shown that the effectiveness of ezetimibe, which blocks NCP1-L1 and inhibits cholesterol absorption, depends on the NCP1-L1 genotype [50].
There are other transporters of cholesterol; for example, SR-B1 is located both in the apical and basolateral membranes of the enterocyte [51]. Scavenger receptors (SR) are cell surface proteins that can bind and internalise modified lipoproteins. SR-B1, which is involved in cholesterol uptake in the intestine and may play an important part in intestinal chylomicron production, and the fatty acid transporter CD36, which is also involved in the uptake of oxidised LDL, are members of the class B scavenger receptor family [52]. Hiashi et al. [53] investigated gene expression of key proteins involved in the active absorption of dietary fat and cholesterol in response to the development of insulin resistance. They used two models of diet-induced insulin resistance, the fructose-fed hamster and the high-fat-fed mouse. Expression of SR-B1 was increased in both the fructose-fed hamster and the high-fat-fed mouse models of insulin resistance. In CaCo2 adenocarcinoma cell line, SR-B1 over-expression increased apo B100 and apo B48 secretion. The authors conclude that apical or basolateral SR-B1 may have an important role in cholesterol absorption and may play a part in cholesterol over-absorption in insulin-resistant states. SR-B1 in the intestine may play an important role in chylomicron production. Cdc42, a member of the Rho family of small guanidine triphosphatases with numerous functions, has been shown by Xia et al. [54] to interact with NCP1-L1 and to control its movement from the endocytic recycling compartment to plasma membranes in a cholesterol-dependent manner. Glucose-stimulated Cdc42 signalling appears to be essential for second stage insulin secretion [55]. It is probable that in insulin resistance the signalling of NCP1-L1 is disturbed through this pathway, but we have been unable to find any studies in the intestine that have explored the pathway in diabetes/insulin resistance. In animal studies we have demonstrated an increase in cholesterol absorption in diabetes [56]. We then asked the question as to whether diabetes might be associated with an increase in cholesterol absorption through stimulation of NCP1-L1. We demonstrated in animal models of diabetes that NCP1-L1 was upregulated [57], and in diabetic patients we demonstrated an increase in NCP1-L1 mRNA [58], suggesting a mechanism for an increase in cholesterol absorption. In the Psammomys obesus, a model of type 2 diabetes, the animals exhibiting weight gain, hyperinsulinaemia and hypercholesterolaemia, NCP1-L1 protein and gene expression were both significantly reduced in the intestine, and the authors found a lower capacity to absorb cholesterol compared to controls [59]. This may suggest interspecies variation, but it is a surprising finding considering that this animal model of diabetes has been shown to have increased production of intestinal apo B48-containing lipoproteins [60]. Ezetimibe has been shown to bind to the brush border and to NCP1-L1 expressing cells [61]. There is a sterol regulatory element in the promoter and a sterol-sensing domain of NCP1-L1 which appears to regulate cholesterol absorption in response to cholesterol intake. Huff et al. [62] have shown that NCP1-L1 is suppressed in mice given a cholesterol-rich diet and increased in the cholesterol-depleted porcine intestine. The nuclear receptor, peroxisome proliferator-activated receptor (PPAR) δ/β, appears to control the expression of NCP1-L1. Activation by a synthetic agonist of PPARδ has been shown to reduce cholesterol absorption and reduce expression of NCP1-L1 without altering expression of the adenosine triphosphate (ATP)-binding membrane cassette transport proteins ABC G5/G8 [63]. ABC G5/G8 is a transmembrane heterodimer that transports plant sterols and excess cholesterol out of jejunal enterocytes (discussed in greater detail below). Fenofibrate, a PPARα agonist, has been shown to inhibit cholesterol absorption; the mechanism has been shown to be through reduced NCP1L1 transcription by binding to a PPARα response element upstream of the human NCP1-L1 gene. In a human construct, Iwayanagi et al. [64] showed that PPARα positively regulated human NCP1-L1 transcription, and Valasek et al. [65] showed that fenofibrate reduced intestinal cholesterol absorption by PPARα modulation of NCP1-L1. Tremblay et al. [66] have shown that atorvastatin increases NCP1-L1 in the intestine and decreased ABC G5/G8 which leads to an increase in cholesterol absorption. These findings were accompanied by an increase in the transcription factors, sterol regulatory element-binding protein (SREBP) 2 and hepatic nuclear factor (HNF)-4.
Intestinal ATP-Binding Cassette Proteins G5/G8
Once cholesterol has been transported across the brush border membrane, it faces another regulatory process and may be excreted back into the intestinal lumen rather than being further processed for absorption into the perimesenteric lymphatic circulation. ABC G5/G8 expression is mostly confined to the human small intestine and liver [67]. These two proteins act in tandem to re-excrete both cholesterol and, in particular, non-cholesterol sterols such as plant sterols from the body. Much of the understanding of ABC G5/G8 comes from the rare mutations that cause a defect in ABC G5/G8 and result in high levels of sitosterol in the blood. Beta-sitosterolemia is a condition which manifests itself in children as tendon xanthomas or in young adults as severe CHD with massive accumulation of sterols and stanols in monocyte-derived macrophages [68]. Ma et al. [69] found in an animal model that dietary calcium had a beneficial effect on lipoprotein profile by upregulating the mRNA levels of intestinal ABC G5/G8 and cholesterol-7α-hydroxylase (CYP7A1), whereas it downregulated the intestinal NCP1-L1 and microsomal triacylglyceride transport protein (MTP) due to enhanced biliary cholesterol excretion. Mendes Gonzales et al. [70] investigated the effect of ABC G5/G8 deficiency on lipoproteins in mice. They found that postprandial triglycerides were fivefold higher in the ABC G5/G8− − mice due to a lower fractional catabolic rate with lower post-heparin lipoprotein lipase activities. They also showed that liver triglyceride secretion and intestinal triglyceride secretion were higher and there was a relationship between this and the HOMA index as a measure of insulin resistance. Since diabetes is so frequently associated with dyslipidaemia and atherosclerosis, the ABC translocases became a target for research. Blocks et al. [71] examined mRNA and protein expression of ABC G5/G8 in the intestine of streptozotocin diabetic rats and found significant reduction in expression of both ABC G5/G8. They found that levels were partially normalised on insulin supplementation. We have shown that ABC G5/G8 were reduced by more than 50 % in the intestine of Zucker diabetic fa/fa rats compared with lean rats although these changes did not reach statistical significance [58]. Insulin treatment caused a nonsignificant increase in ABC G5/G8 mRNA. In another study of streptozotocin diabetic rats, ABC G5/G8 were both very significantly reduced in the intestine [57]. There was a negative correlation between ABC G5/G8 and chylomicron cholesterol [57]. In the Psammomys obesus, another model of diabetes, Levy et al. [59, 60] showed a reduction in ABC G5/G8 in the intestine. In the intestine of human subjects with type 2 diabetes, ABC G5/G8 mRNA were both significantly lower compared to controls [58]. There was a negative correlation between ABC G5/G8 and NCP1L1 in the combined diabetic and control subjects, and there was a significant negative correlation between chylomicron cholesterol and both ABC G5/G8 [57]. These two genes appear to play an important role in the dysregulation of cholesterol metabolism in diabetes.
Microsomal Triglyceride Transport Protein
The cholesterol that has evaded ABC G5/G8 in the intestine is now ready to be solubilised for transport through the lymphatic system. The assembly of the chylomicron occurs under the direction of microsomal triglyceride transfer protein (MTP). MTP has the ability to combine cholesterol, triglyceride and phospholipid into the triglyceride-rich chylomicron particle. The cholesterol that becomes available however not only is cholesterol that has been absorbed from the diet but is also cholesterol that has been excreted through the bile duct under the influence of hepatic ABC G5/G8. Finally, there is the cholesterol that has been synthesised in the intestine through the 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) reductase pathway. This pathway in the intestine accounts for up to 25 % of body-synthesised cholesterol, the amount varying depending on whether the patients are high or low cholesterol absorbers. Intestinal MTP plays a major role in the assembly of the chylomicron particle and therefore of cholesterol and triglyceride metabolism. MTP has become a hot topic since inhibitors of intestinal MTP have been shown to lower triglyceride without causing hepatic steatosis at least in animal studies [72, 73]. In short-term human studies, a specific intestinal MTP inhibitor did not appear to effect liver function tests [74]. Although many polymorphisms of MTP have been described, some of which have considerable impact on LDL cholesterol in both nondiabetic and diabetic subjects, it is difficult to know whether the results mainly stemmed from the effect in the liver rather than the intestine [75, 76]. The intestinal inhibitors of MTP which have no effect on the liver should answer this question in the future. In animal studies, diabetes is associated with an increase in MTP mRNA with close correlation between MTP mRNA and chylomicron cholesterol [77–80]. In the diabetic rabbit, increased intestinal MTP mRNA is associated with an increase in chylomicron particle numbers [77], but in the rat it is associated with larger particles [78]. The fructose-fed insulin-resistant hamster model had an increase in MTP protein mass, and this was associated with an increase in the triglyceride-rich intestinally derived lipoproteins [79]. Zolotowska et al. [80] in 2003 [80] examined the B48-containing lipoprotein assembly in the small intestine of Psammomys obesus, a model of nutritionally induced diabetes and insulin resistance. De novo triglyceride synthesis, apo B48 biogenesis and triglyceride-rich lipoprotein assembly were all increased. MTP activity and protein expression, however, were not altered. In the enterocyte of fructose-fed golden hamster, MTP mRNA and protein mass were increased by TNFα, but apo B levels in the enterocyte were not effected suggesting that there is considerable interspecies variation [80]. In humans with type 2 diabetes, we demonstrated an increase in MTP mRNA in intestinal biopsies [58, 81]. Diabetic patients who were on statin therapy had lower MTP mRNA compared to those not on statins [81]. We found positive correlations between MTP mRNA and chylomicron fraction cholesterol and apo B48 [81]. A novel intestinal-specific inhibitor of MTP has been shown to ameliorate impaired glucose and lipid metabolism in Zucker diabetic fatty rats, but whether this effect was due to impairment of food intake or to inhibition of fat absorption is not clear [82].
The signals that upregulate chylomicron formation to cope with excess fat in the diet are slowly being elucidated. Another non-specific inhibitor of MTP, which reduced serum levels of triglycerides by more than 70 %, was also associated with significant improvements in glucose tolerance and insulin sensitivity in Zucker fatty rats [83]. Hepatic MTP mRNA expression is negatively regulated by insulin, and it is suggested that insulin might also directly inhibit apo B48 secretion independently of MTP even though it is probable that upregulation of MTP stimulates apo B secretion [84]. The membrane glycoprotein CD36 binds long-chain fatty acids. CD36 deficiency reduces chylomicron production [84]. It has recently been shown that binding of lipid by CD36 upregulates apo B48 and MTP through CD36 signalling via the ERK-1/ERK-2 pathway [85]. Interestingly polymorphisms of MTP which have been associated with differences in serum lipids appear to alter cholesterol absorption but not synthesis in women [86].
Apolipoprotein B48 and B100
Apo B48, the structural protein for the chylomicron, is produced in the intestine by editing of the hepatic version, apo B100 [87]. The enzyme apobec cuts the apo B100 form into the shorter version, apo B48. It has been suggested that apo B is in excess of body needs. In the liver recent work has demonstrated that insulin silences apo B translation by introducing intracellular traffic into mRNA granules [88]. The authors showed that the availability of apo B mRNA for translation was regulated by the rate of release from translationally silenced mRNPs processing bodies (p bodies). Insulin specifically silences apo B mRNA translation by reprogramming its mRNA into p bodies and reducing the size of translationally competent mRNA pools. Translational control via traffic into cytoplasmic RNA granules may be an important mechanism for controlling the rate of apo B synthesis and hepatic lipoprotein production, the authors suggest. It is however not clear that this silencing plays a part in reducing chylomicron production or influences nascent chylomicron size. In diabetes it may be that there is an increase in apo B48 production, but then if meaningful, one would expect smaller chylomicron particles containing less triglyceride per particle to be produced. Our studies in an animal model demonstrated that the particles in the cannulated lymphatic duct of the rabbit were associated with an increase in chylomicron particle numbers [89], but in the rat it was associated with larger particles [56]. In patients with type 2 diabetes, apo B48 is increased, but it is difficult to ascertain whether the increase is due to delayed delipidation, increased synthesis or both [90]. We injected labelled chylomicrons, collected by cannulation of the lymph duct into diabetic and nondiabetic rabbits, into another group of diabetic and nondiabetic rabbits and found evidence of both increased synthesis and delayed clearance [89]. Our animal research therefore suggests that the increase in apo B48 particles in diabetes may be due to both an increase in synthesis and a decrease in turnover.
Cholesterol Synthesis and HMGCoA Reductase
Cholesterol synthesis is regulated by HMGCoA reductase, the rate-limiting enzyme in the synthetic pathway. Sterol regulatory element-binding protein-2 (SREBP-2) is a regulatory protein of cholesterol homeostasis and regulates HMGCoA gene expression. In isolated rat hepatocytes, we demonstrated significant reduction in HMGCoA reductase activity in the presence of insulin [91]. In animal studies we have recently reported the different effects of pioglitazone, an insulin sensitizer which acts through peroxisome proliferator-activated receptor (PPAR) gamma, as compared to insulin on expression of hepatic HMGCoA reductase mRNA [42]. We found a highly significant increase in expression of HMGCoA reductase in the liver of diabetic animals (Zucker diabetic fatty fa/fa rats). There was a small but insignificant reduction in HMGCoA reductase mRNA in the intestine when the animals were treated with insulin [42]. There was a larger reduction in HMGCoA reductase in the liver of the insulin-treated animals, but this reduction did not reach statistical significance [42]. In type 1 diabetes Sittiwet et al. [92] presented some evidence to suggest that improved glycaemic control increases cholesterol synthesis. However, the study was perhaps a little unsatisfactory in that, although there was a significant increase in cholesterol synthesis, there was no change in serum or lipoprotein cholesterol nor was there any change in markers of cholesterol absorption. Inhibition of HMGCoA reductase with a statin has been shown to decrease ABC G5/G8 as well as increasing NCP1L1, thus increasing cholesterol absorption [81].
Hepatic steatosis is common in diabetes, insulin resistance and obesity. Inflammatory stress is present in these conditions and is also associated with obesity, insulin resistance and diabetes. It is therefore of interest that Zhao et al. [93] demonstrated that interleukin 1b and interleukin 6 stimulation of Hep G2 cells increased SREBP2 and HMGCoA mRNA. Further high-fat loading in mice or LDL loading in Hep G2 cells suppressed the above genes, but this suppression could be overridden by the above inflammatory proteins [93]. Severe calorie restriction in patients with steatosis results in rapid reduction of liver fat, insulin resistance and improvement in diabetes control. In contrast, insulin resistance and the accompanying hyperinsulinaemia are associated with an upregulation of SREPB2 through extracellular signal-regulated pathways involving the kinases ERK-1 and ERK-2, another example of the interaction between fat and carbohydrate metabolism [94] (for review, see Van Rooyen and Farrell [95]).
Very-Low-Density Lipoprotein
Before discussing clearance of the chylomicron particle with reference to insulin resistance and diabetes, it is necessary to discuss VLDL, the other major triglyceride transport particle which is produced by the liver and has as its structural protein apo B100. The synthesis of the VLDL particle in the liver is somewhat similar to that of the chylomicron in the intestine. Through a series of steps, the lipid and cholesterol are assembled under the influence of MTP with apo B100 yielding VLDL. The VLDL particle will contain some de novo synthesized cholesterol.
As with the chylomicron, apo E attaches itself to the particle and is necessary for clearance by the liver through the LDL B/E receptor. The VLDL particle is distinguished from the LDL particle, not only by its triglyceride content which is much higher than LDL, but also by the attachment of apo E onto the particle. There are three common polymorphisms of apo E. Apo E2/2, although rare, is associated with hypercholesterolemia but E4/4 with hypertriglyceridemia [96]. Compared with individuals with the E3/3 genotype, E2 carriers have a 20 % lower risk of coronary heart disease, and E4 carriers have a slightly higher risk [97]. It has been suggested that the apo E4 allele is a risk factor for the metabolic syndrome [98]. ApoC-III can be present on apo B-containing lipoproteins but is not integral to the basic lipoprotein particle structure; thus, lipoproteins exist both with and without apoC-III. Apo B-containing lipoproteins with apoC-III are enriched in triglyceride and cholesterol and have slow clearance from plasma. The concentration of apoC-III in VLDL and LDL is highly and independently predictive of coronary heart disease, more so than triglyceride alone [99]. LDL particles with apoC-III, a remnant particle produced by partial lipolysis in plasma of VLDL [100], is the lipoprotein particle type most predictive of CVD in type 2 diabetes [101]. ApoC-III inhibits lipoprotein lipase and triglyceride hydrolysis as well as direct clearance of VLDL particles from plasma, resulting in the formation of less LDL.
In passing one might mention apo A5, a key gene regulating triglyceride levels and was thought to be exclusively in the liver. Recently Guardiola et al. [102] have described the expression of the gene in the mouse and human small intestine. The function here has yet to be explained. Dallinger Thie et al. [103] examined apo A5 in diabetes in relation to triglycerides and found the same positive relationship between apo A5 as in non-diabetic subjects. They found in a group of 215 subjects with type 2 diabetes taken from the Diabetes Atorvastatin Intervention Study that 6 % of the variation in plasma triglycerides was due to apo A5 whereas 52 % was explained by apoC-III. Diabetes sometimes results from pancreatitis which may be caused by severe hypertriglyceridemia. Apo A5 has not been shown to play a part in diabetes secondary to pancreatitis [104].
Cholesterol Synthesis and Transport in the Liver
Cholesterol either may be synthesized in the liver through the HMGCoA reductase pathway and packaged for transport by association with apo B100 or may have been delivered to the liver by the chylomicron particle. Insulin plays a major part in regulating many of the steps in the production of cholesterol [105]. HMGCoA reductase is increased in animal models of diabetes in the liver [91]. In isolated rat hepatocytes, we have demonstrated significant reduction in HMGCoA reductase activity in the presence of insulin [106]. In animal studies we have more recently reported the different effects of pioglitazone, an insulin sensitiser through peroxisome proliferator-activated receptor (PPAR) gamma, as compared to insulin on expression of intestinal and hepatic HMGCoA reductase mRNA [42]. In that study we also found a highly significant increase in expression of HMGCoA reductase in the liver of diabetic animals (Zucker diabetic fatty fa/fa rats). There was a large reduction in HMGCoA reductase in the liver of insulin-treated animals, but this reduction did not reach statistical significance (116 + 122 vs. 143 + 48 arbitrary units n + 10) [42]. The liver, like the intestine, can regulate, at least to some extent, the amount of cholesterol in the VLDL particle by regulating the excretion of cholesterol in bile.
Hepatic NCP1-L1
NCP1-L1 is localised to the canalicular membrane in hepatocytes where it plays a part in the regulation of cholesterol transport. Hepatic nuclear factor-1 (HNF-1) alpha and sterol regulatory element-binding protein-2 (SREBP-2) appear to be important regulators of NCP1-L1 in the liver [107]. It has also been shown that they have important binding sites within the human NCP1-L1 promoter. The role of NCP1-L1 in the liver is probably to divert cholesterol away from excretion in the bile [108]. A recent study in female Chinese women with gallstones has shown reduced NCP1-L1 mRNA and protein in the liver and supersaturation of cholesterol in the bile [109]. Ezetimibe has not been shown to increase the risk of gallstones perhaps because the drug has its primary effect in reducing cholesterol absorption. Indeed in the golden Syrian hamster, ezetimibe reduced the diet-induced increase in biliary cholesterol [110], and in gallstone-susceptible mice fed lithogenic diets, ezetimibe prevented gallstone formation [111]. Inhibition of NCP1-L1 by ezetimibe is associated with an improvement in hepatic steatosis. Jia et al. [112] investigated the mechanism by deleting NCP1-L1 in mice and inducing hepatic steatosis with a high-fat diet. The knockout mice did not develop steatosis. Hepatic fatty acid synthesis and mRNA for genes regulating lipogenesis were reduced and the knockout animals did not develop hyperinsulinaemia. Nomura et al. [113] demonstrated in Zucker rats that ezetimibe improved hepatic insulin signalling as well as hepatic steatosis both in the liver and in cultured steatotic hepatocytes. The drug recovered insulin-induced Akt activation and reduced gluconeogenic genes. The relevance of this study to humans is not clear as patients with diabetes who are treated with ezetimibe do not improve blood sugar control [114]. Over-expression of NCP1-L1 in the liver inhibits biliary cholesterol secretion and raises serum cholesterol suggesting that inhibitors of NCP1-L1 may have a role in the liver [115]. NCP1-L1 mRNA has been shown to be increased in the liver of diabetic rats with a positive correlation between NCP1-L1 mRNA and VLDL cholesterol [57]. Interestingly, we found that although pioglitazone significantly reduced NCP1-L1 in the liver of diabetic fa/fa rats, insulin had no effect [42]. ABC G5/G8 play a role in the regulation of cholesterol excretion by promoting excretion of neutral steroids into the bile. Diet-induced lipid loading into the liver causes a significant increase in the expression of ABC G5/G8 in the bile canaliculi [116].
Hepatic ABC G5/G8
The liver X receptor (LXR) helps to regulate expression of ABC G5/G8 [117], and ABC G5/G8 expression is also stimulated by LXR agonists but not to the same extent as by feeding. It has recently been demonstrated that NCP2, a cholesterol-binding protein secreted by the biliary system, positively regulates biliary secretion of cholesterol through stimulation of ABC G5/G8. It is well documented that there is a relationship between gallstones, diabetes and the metabolic syndrome. Under conditions of obesity and insulin resistance, the serine/threonine protein kinase Akt/PKB is required for lipid accumulation in liver. Two forkhead transcription factors, FOXA2 and O1, have been suggested to function downstream of, and to be negatively regulated by, Akt and are proposed as key determinants of hepatic triglyceride content [118]. In mice with isolated insulin resistance, there was an increased expression of biliary transporter ABC G5/G8 through the disinhibition of the forkhead transcription factor FOX 01 [119]. However, these findings do not fit well with the increased VLDL synthesis that has been described in insulin resistance and diabetes [120–122] even if they explain the increased gallstones found in diabetes [123, 124]. In diabetic fa/fa rats insulin but not pioglitazone significantly increased hepatic expression of ABC G5/G8 [42].
MTP in the Liver
The final stage for the production of the VLDL particle is the assembly of the cholesterol, triglyceride and phospholipid with apo B100 under the regulation of MTP. In animal studies MTP mRNA is increased in streptozotocin diabetic rats in the liver [57]. We have shown modest suppression with insulin treatment in the liver of Zucker diabetic rats [42]. In the alloxan diabetic rabbit model, we found no difference in MTP mRNA or activity in the liver, whereas there was a significant increase in the intestine [77]. The disturbances in chylomicron and VLDL production reflect the increase in chylomicron and VLDL particles found in diabetes. Although chylomicron production has only been measured in animal models of diabetes and insulin resistance, VLDL overproduction has been shown in diabetes in humans as well [120–122, 125], the driving force being the non-supressability of FFA postprandially due to the malfunction of adipose tissue by adipose triglyceride lipase (ATGL) leading to an increase in postprandial triglycerides which are taken up by the liver and re-secreted in the VLDL [126]. VLDL overproduction is also found in insulin resistance [127].
Triglyceride-Rich Lipoproteins in Diabetes
One of the causes of the disturbance in the metabolism of the triglyceride-rich lipoproteins in diabetes is the inhibition of lipoprotein lipase (LL) activity. Lipoprotein lipase is an insulin-sensitive enzyme. In type 2 diabetes insulin treatment significantly increases LPL activity in adipose tissue [128, 129]. Among the causes of lipoprotein lipase dysfunction is the effect of glucose on enzyme dimerization and has been related to the severity of diabetes [130]. Clearance of both VLDL and chylomicrons is severely impeded due to the inability of lipoprotein lipase to function in a relative or absolute insulin-deficient environment. The VLDL remnant is cleared by the LDL B/E receptor or the VLDL lipoprotein receptor (VLDLr) which is found mainly in the adipocyte and muscle cells [131]. The LDL receptor-related protein which clears postprandial lipoproteins is also insulin dependent [132]. Perhaps one of the most important results of this delay in clearance is the influence that the triglyceride-rich lipoproteins have on LDL size and atherogenicity [133]. Large triglyceride-rich VLDL is associated with both increased small dense LDL and reductions in HDL.
LDL in Diabetes
The concentration of LDL is often normal in diabetic when compared to non-diabetic patients of similar BMI. However, since turnover of the LDL particle is increased in diabetes [134] and production is increased through the increase in hepatic VLDL secretion, a similar serum level of LDL in diabetes should not be taken as meaning that the atherogenic risk of LDL should be presumed to be similar to that in the non-diabetic and is one of the reasons why emphasis is placed on maximum reduction of LDL in diabetes. HMGCoA reductase and the LDL receptor are insulin sensitive and the receptor is down-regulated and HMGCoA reductase is up-regulated in the setting of insulin resistance [135]. As stated previously, small dense LDL is more common in diabetes [133], and small dense LDL has been shown to promote macrophage foam cell formation [136]. LDL composition is abnormal in other ways, i.e. it contains more esterified cholesterol [137], an increase in linoleic acid and more FFA [13]. However, a more recent study suggests that patients with diabetes and the metabolic syndrome have lower cholesterol ester and lower linoleic acid in the cholesteryl esters [138]. This may of course be due to differences in diets between the 2 studies. Both studies agree that markers of lipid peroxidation are increased in diabetes. Colas et al. [138] have also shown that LDL from metabolic syndrome and type 2 diabetic patients was potent in activating the platelet arachadonic acid-signalling cascade, potentiating platelet aggregation as compared to control LDL. An increase of free radical production occurs in hyperglycemia of diabetes [139] causing LDL glycation, and glycated LDL is more susceptible to oxidation, one reason for the increased LDL oxidation that occurs in poorly controlled diabetes [140]. The foam cell is perhaps the hallmark of the atherosclerotic lesion. The development of the foam cell depends on macrophage uptake of cholesterol. A novel mechanism has recently been described which facilitated uptake of oxidised LDL through uptake by the toll-like receptor 4 (TR4) which is found on the macrophage surface [141, 142]. It has been demonstrated that cholesterol ester hydroperoxides are an indigenous ligand for TR4. The increase in free fatty acids in poorly controlled diabetes is associated with an increase in fatty acids attached to the LDL particle, a further potential cause for increased oxidation of the particle [13]. Oxidized LDL can be taken up by the macrophage through scavenger receptors in an unregulated manner. We have shown that lipoprotein lipase mass is increased on diabetic LDL [13], a factor which also increases LDL uptake into plaque (Fig. 5.5).
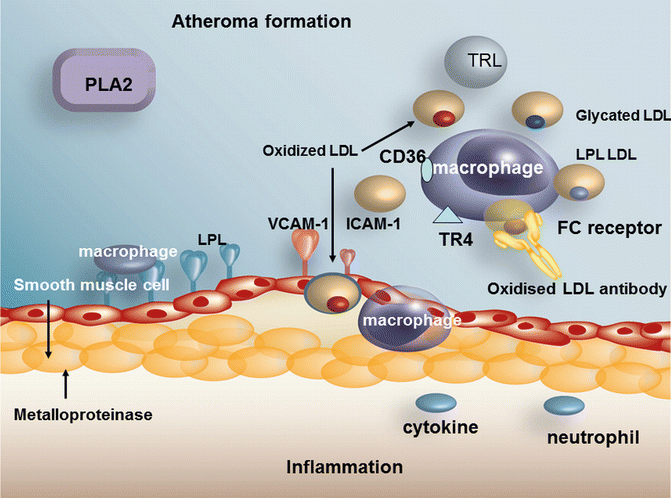
 Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Production and Metabolism of Triglyceride-Rich Lipoproteins in Both the Normal and Diabetic States
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Production and Metabolism of Triglyceride-Rich Lipoproteins in Both the Normal and Diabetic States
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus




Related posts:
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes
Production and Metabolism of Triglyceride-Rich Lipoproteins in Both the Normal and Diabetic States
Cholesterol Absorption Inhibitors (Ezetimibe) and Bile Acid Binding Resins (Colesevelam HCl) as Therapy for Dyslipidemia in Patients with Diabetes Mellitus
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
