Fig. 7.1
Schematic view of Lp(a)
Table 7.1
Average composition of Lp(a) in comparison to LDL
Lp(a) (%) | LDL (%) | |
|---|---|---|
Protein | 30.0 | 21.0 |
Carbohydrates | 10.0 | 1.3 |
Cholesteryl esters | 31.5 | 42.0 |
Free cholesterol | 7.0 | 9.0 |
Phospholipids | 16.0 | 20.7 |
Triglycerides | 5.5 | 6.0 |
Apo(a), the characteristic glycoprotein component of Lp(a), has a unique structure. It consists of repetitive protein segments, so called kringles (K) that are highly homologous to the K-four (K-IV) in plasminogen. K-IV contain approx. 110 amino acids forming a secondary structure, which resembles “Danish kringles” [4]. The N-terminal part of apo(a) consists of numerous repetitive copies of these kringle-IVs. Apo(a) in addition has one copy of a K-V-like kringle and a protease-like domain similar to plasminogen. In humans, there exist probably 30 and more genetically determined apo(a) isoforms, giving rise to a great size heterogeneity. The smallest apo(a) isoform contains the protease domain, one copy of K-V and 11 K-IVs of which K-IV Type 1 (T-1) and T-(3–10) are unique in their primary structure, whereas K-IV T-2 is present in two identical copies. Larger isoforms differ by the number of K-IV T-2s; the largest apo(a) described so far had 52–54 K-IVs. Between the K-IV domains, there are linker regions, which are highly glycosylated by N– and O-linked sugars. Although the majority of apo(a) is complexed to LDL, there are small and variable amounts, which are present in plasma in the free form [5] and found in the bottom fraction after ultracentrifugation. Free apo(a) is prone to proteolytic degradation, and the generated fragments are secreted into urine (see below).
Lp(a) Metabolism
Assembly of Lp(a)
Lp(a) is biosynthesized only in humans and old world monkeys which poses some problems in studying its metabolism in detail. Apo(a) expression takes place primarily in the liver, yet small amounts of APOA mRNA have also been detected in testis and brain. Their role on the overall Lp(a) metabolism is unknown. Hepatocytes from primates have been found to synthesize a preform of apo(a) with a lower degree of glycosylation. Upon maturation, intracellular apo(a) reaches the Golgi apparatus and is secreted in mature form as a glycoprotein most probably without attached LDL. The genetically determined size of apo(a) reflecting the number of K-IV repeats correlates with the intracellular residence time, and thus, small isoforms are secreted much faster as compared to large isoforms. This appears to be the reason for the existence of the negative correlation between apo(a) size and plasma Lp(a) concentration. We and others have found that the assembly of Lp(a) from apo(a) and LDL is a two-step process [6]. In the first step, specific K-IVs of apo(a), mostly K-IV T3-6, bind non-covalently to Lys groups on apoB of LDL. This binding is reversible and may be dissociated by Lys analogues such as epsilon aminocaproic acid, tranexamic acid, and others [7]. It has been argued that by interfering with the first step of Lp(a) assembly, plasma Lp(a) levels may be reduced, as free apo(a) is degraded faster than LDL-bound apo(a). In the in vivo and in vitro experiments, however, this assumption has been refuted, and plasma apo(a) and Lp(a) levels went in opposite directions, i.e., were twofold and higher elevated [7]. The reason for these observations may be explained by the fact that free apo(a) binds to the surface of liver cells and upon contact with LDL forms the first Lp(a) complex. Apo(a) which does not find its way to LDL is internalized and degraded. We actually could demonstrate that cell bound apo(a) dissociates upon treatment with Lys analogues and assembles with LDL more efficiently.
When recombinant apo(a) was mixed with LDL in vitro, a fast assembly took place, characterized by stable disulfide bridging. This mechanism that most probably also occurs in vivo does not need any enzymatic activity and leads to the formation of the final mature Lp(a). Interestingly, apo(a) has a preponderance for binding apoB100 from humans and few animal species, yet apoB100 from rodents, in particular from mice, hardly form any Lp(a) upon incubation with apo(a). Thus, the metabolism of Lp(a) might be studied only in double transgenic human apo(a):apoB100 mice or in monkeys.
Although the evidence for an extracellular assembly is favored by many investigators, there are still experimental data that may be only interpreted if an intracellular assembly of Lp(a) is assumed [8]. Further experiments will be needed to reach a definite conclusion.
In Vivo Metabolism of Lp(a)
As common laboratory animals do not express apo(a), the Lp(a) metabolism had to be studied in humans. We actually were the first to demonstrate that plasma Lp(a) concentrations highly significantly correlate with the production rate, yet the Lp(a) catabolism does not control Lp(a) levels [9]. This is demonstrated in Fig. 7.2, where we studied 9 probands with Lp(a) levels ranging from 5 to 75 mg/dl. Our results have been confirmed subsequently by other investigators using radioactive Lp(a) tracers or stable isotope precursors [10].
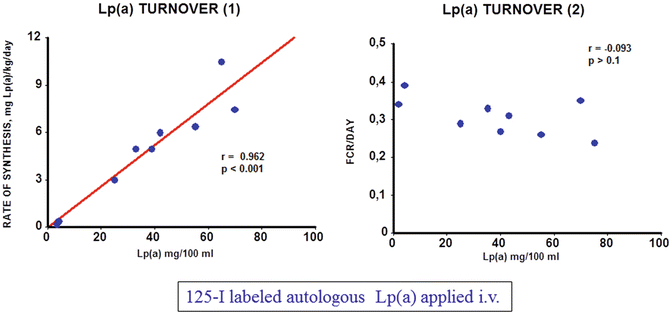
Fig. 7.2
Turnover of Lp(a) in humans. 125I radiolabeled Lp(a) was injected into 9 volunteers with plasma concentrations ranging from 5 to 75 mg/dl and the decay of the specific radioactivity was followed over time (details are found in [9])
Concerning the catabolism of Lp(a), the liver appears to be the major organ of Lp(a) degradation. This has been proven by turnover studies in several animal species including rats, rabbits, mice, and hedgehogs. The latter animal model has been chosen since it synthesizes a lipoprotein that resembles Lp(a) [11]. In the in vivo studies, we found that approx. 50 % of Lp(a) is taken up by the liver, followed by kidney, spleen, and muscle. Unfortunately, the mechanism of in vivo cellular uptake of Lp(a) is mostly unknown. In the in vitro experiments, Lp(a) has been shown to bind to the LDL receptor—yet with a greatly reduced affinity as compared to LDL. There have been also reports suggesting an in vitro binding of Lp(a) to other receptors such as VLDL receptor, Gp-330 (megalin), asialoglycoprotein receptor, and scavenger receptors, yet their relevance for the in vivo metabolism remains to be established.
Regulation of Apo(a) Transcription
The transcription of genes involved in lipid and lipoprotein metabolism is heavily influenced by nuclear receptors including PPARs, RXR, CAR, PXR, LXR, and FXR, in addition to others [12]. These nuclear receptors in concert with other transcription factors coordinate pathways involved in lipid absorption, de novo biosynthesis, cell excretion from different organs, and conversion of cholesterol to steroid hormones and bile acids. In the bile acid metabolism, FXR is of major importance as it controls for the overproduction of bile acids and detoxification of liver and cells from the biliary tract. FXR is also involved in glucose homeostasis, intestinal bacterial infection, and tumorigenesis of liver [13]. Although the molecular mechanism of FXR action is not elucidated in full detail, one may say the following: FXR is mainly expressed in the liver, intestine, kidney, and adrenals and binds to response elements in the promoter as heterodimer together with RXR, thereby transactivating or transrepressing cognate target genes. Of key importance is the transactivation of small heterodimer partner (SHP) and of FGF-15/19. SHP is a transcriptional repressor that has no DNA binding capability but rather interacts with the DNA binding and/or activating factor domain of numerous nuclear receptors, among them HNF4, LRH-1, estrogen receptor (ER), and RXR, thereby interfering with gene transcription [14]. Mouse FGF-15 that is expressed almost exclusively in the intestine and its human orthologue FGF-19 expressed in the small intestine but also in the liver are also transactivated by FXR-RXR heterodimers. It has been demonstrated that FGF-15/19 signals from intestine to liver to repress the transcription of key enzymes of bile acid biosynthesis. Inagaki et al. [15], for example, provided evidence that FGF-15 represses CYP7A1 in wild-type mice but did not affect CYP7A1 mRNA levels in SHP−/− mice. It was suggested that in the FGF15/19 pathway, post-transcriptional activation of SHP may take place. It was also shown that FGF binding to its receptor, FGFR4, signals via the MAP-kinase pathway, thereby interfering with CYP7A1 transcription [16]. In a recent report, Song et al. [17] published that the MAPK-ERK1/2 pathway is a major trigger of FGF19-mediated inhibition of CYP7A1. Binding of FGF19 to FGF4R led to Tyr phosphorylation of the latter and in turn to a phosphorylation cascade of RAS, c-RAF, MEK1/2, and MAPK/ERK1/2 and finally to the transcriptional inhibition of CYP7A1. This pathway was independent of SHP.
In our recent studies, we made the observations that patients suffering from obstructive cholestasis with high plasma bile acid concentrations had comparatively low plasma Lp(a) levels. When patients were treated by surgery, plasma bile acid levels normalized, and plasma Lp(a) rose significantly to levels compatible with their individual isoform [18] (Table 7.2). This led us to hypothesize that FXR might be responsible for these observed changes in plasma Lp(a). We therefore performed a serious in vivo studies with transgenic mice expressing apo(a) under the control of its native human promoter, in addition to in vitro studies using cultured primary hepatocytes from these mice, aimed at elucidating the role of FXR ligands in apo(a) transcription. In a first report published in JCI [18], we show that the apo(a) promoter contains at nucleotide 814–826 upstream to the transcription initiation site, a direct repeat (DR-1) that binds HNF4a with high affinity, thereby transactivating apo(a) transcription. FXR upon activation by bile acids or synthetic ligands is transported from the cytosol to the nucleus and competes with the HNF4a binding to the DR-1, thereby downregulating apo(a) transcription. This pathway was further proven by reporter assays using a 2kB promoter sequence of apo(a) in front of the luciferase gene.
Table 7.2
Influence of drugs and other substances on plasma Lp(a) concentrations
Substance | % Decrease |
|---|---|
Omega-3 fatty acids | 5–20 |
Palm oil | 10–25 |
Vegetarian diet | 10 |
Nicotinic acid and derivatives | 15–35 |
Aspirin | 15–20 |
l-carnitine | 10–15 |
Lp(a)/LDL apheresis | 60–80 |
ACE inhibitors | 10–40 |
Feeding mice with a diet containing 0.2 % cholic acid, the mouse ligand for FXR, led to a reduction of plasma apo(a) to almost zero. The pathway described above however accounted for only approx. 60 % of the downregulation of apo(a) transcription. We therefore performed additional experiments exploring the possibility that SHP and/or FGF-15/19 might be responsible for the remaining 40 % repression of apo(a) transcription. Overexpression of SHP in primary hepatocytes from transgenic apo(a) mice did not downregulate apo(a) biosynthesis. These findings were backed up also by promoter studies using a luciferase reporter assay. On the other hand, we could show that the addition of FGF-19 to primary hepatocytes of the apo(a) transgenic mice downregulated apo(a) transcription and protein biosynthesis. Knockdown of the FGF-15/19 receptor (FGFR4) on the primary hepatocytes by a specific siRNA abolished the effect of FGF15/19. We also could prove that in this pathway, FGF15/19 binding to FGFR4 activates a phosphorylation cascade involving RAS-RAF-MEK1/2 ERK1/2 that leads to ELK-1 phosphorylation and translocation to the nucleus. Using luciferase reporter assays in combination with site directed mutagenesis, we finally identified an ETS domain at nt −1,615 to −1,630 that was responsible for P-ELK-1 binding and repression of APOA transcription [19]. These two pathways are schematically displayed in Fig. 7.3. We believe that the clarification of these pathways may serve as basis for developing new medications to treat individuals with elevated plasma Lp(a) levels that are at high risk for CAD and MI.
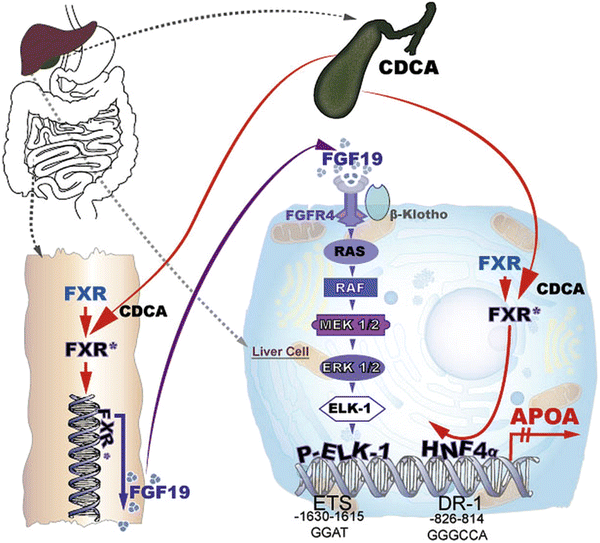
Fig. 7.3
Cartoon of the transcriptional regulation of apo(a)
Although bile acids are capable in downregulating apo(a) transcription almost completely by the two pathways described above, there are numerous additional regulatory sequences in the apo(a) promoter that need to be addressed in future investigations.
Genetics of Lp(a)
Lp(a) concentrations in human plasma range from <1 mg/dl to >250 mg/dl and are to >90 % genetically determined. The gene for apo(a) is located on chromosome 6q26–q27. Utermann et al. were first to recognize that apo(a) shows a tremendous size heterogeneity that is based on the number of K-IV repeats [20]. This size heterogeneity correlates with plasma Lp(a) levels. Large isoforms were found to correlate with low plasma Lp(a) levels and vice versa. The molecular mechanism of these findings is based on one hand on the fact that large apo(a) isoforms are trapped and degraded during biosynthesis in the rough endoplasmatic reticulum and in the Golgi compartment to a much greater extent than small isoforms [21].
The promoter region of apo(a) contains a variable number of tandem repeats (VNTR) with the pentanucleotide TTTTA in addition to the +93 C/T polymorphism of the untranslated region in the apo(a) gene. Further mutations and polymorphisms are abundant in the apo(a) gene that explain many but not all variations in plasma Lp(a) levels. Ichinose [22], for example, identified two additional functional SNPs in the distal enhancer region 20 kB upstream of the apo(a) gene. In addition, numerous polymorphisms were identified in the K-IV domains of the apo(a) gene that showed significant impacts on apo(a) plasma concentrations. As mentioned above, in the proximal apo(a) promoter region contains numerous regulatory sequences including binding sites for HNF1 and HNF4, IL-6, SREBPs, CREB, and many more. It will be interesting to study the influence of genetic variations in these transcription factors in addition to their DNA binding sites in apo(a) in view of their impact on genetically determined variations of plasma Lp(a) levels.
Another point that needs attention is the fact that there exist great ethnic differences in the distribution of plasma Lp(a) levels. Africans and African Americans, for example, have much higher Lp(a) levels than the white population, considering the individual size polymorphism. The opposite is the case with Chinese individuals and other Asian populations. The reason for these differences has never been explored at a molecular basis and will need much more attention in future.
Factors Affecting Plasma Lp(a)
There are numerous factors that have been described to permanently or transiently modulate plasma Lp(a) concentrations [23]. A comprehensive list of most of these factors is shown in Table 7.3.
Table 7.3
Factors affecting plasma Lp(a) concentrations
Genes and diseases | ||
Genes | Effect | |
APOA | Size polymorphism (50 %); other 40 % | |
APOE | Variable | |
LDL-R | Increase | |
MODY (HNF1/4a) | Variable | |
Diseases | ||
Acute phase | Increase | |
Renal disease | Increase | |
Diabetes mellitus | Increase | |
Cancer: different forms | Increase | |
Gout | Increase | |
Antiphospholipid antibodies | Increase | |
Liver disease | Decrease | |
Hyperthyroidism | Decrease | |
Hypothyroidism | Increase | |
Others | ||
Alcohol | Decrease | |
Menopause | Increase | |
Hormones | ||
Compound | Effect | Comment |
Estrogens | Up to 35 % reduction | Rebound effects have been observed |
Progesterone | 30–40 % reduction | Only few studies reported |
Tamoxifen | 35 % reduction | Antiestrogen |
Tibolone | 35 % reduction | Synthetic steroid hormone |
Raloxifene | 18 % reduction | Estrogen-R modulator |
Testosterone | 30–40 % reduction | Only short observations |
Pregnancy | Two-fold increase | Normalized postpartum |
Anabolic steroids | 60–70 % reduction | Not for clinical use |
ACTH | 30–40 % reduction | Few observations |
Conventional drugs | ||
Compound | Effect | Comment |
Niacin | 20–30 % reduction | Currently most recommended |
Fibrates | <20 % reduction | Large study with gemfibrozil |
Statins | Inconsistent | Significant increases in Lp(a) were reported |
Neomycin (2 g/d) | 24 % decrease | Interferes with release of apo(a) from liver cell surface |
N-act-cys | Controversial | Antioxidant, reduces –S–S– bonds |
l-carnitine | 10–20 % decrease | Mitochondrial FA transport |
ASA | 10–20 % reduction | Even at low dose efficient |
Apheresis | 50–80 % reduction | Independent of the system except for AB column |
New medications under investigation | ||
Compound | Effect on Lp(a) | Mechanism |
Mipomersen | >30 % reduction | siRNA against apoB |
Eprotirome | Up to 40 % reduction | Thyroid mimetic |
PSK-9 inhibitor | ? | PSK-9 antibody |
Lomitapide | ? | MTP inhibitor |
Anacetrapib | 30–50 % reduction | CETP inhibitor |
Different factors | ||
HGH treatment | 2× increase | |
Smoking | 10–20 % reduction | |
Obesity | 10–20 % reduction | |
Omega-3 FA | 10–20 % reduction | |
Stearic acid | Up to 25 % increase | |
Trans-FA | Up to 25 % increase | |
Conjugated linoleic acid | Up to 25 % increase | |
Cyclosporine | 2× increase | |
In addition to the apo(a) gene, other genes involved in lipid metabolism such as apoE, LDL-R, and HNF1 and HNF4 have variable effects. Among the secondary factors, renal and liver diseases appear to be the most striking ones. In kidney diseases, Lp(a) is elevated two- to three-fold, and it appears that nephrotic syndrome and end-stage renal disease have a different etiology for elevating Lp(a). While in nephrotic syndrome the rate of Lp(a) biosynthesis was found to be increased, end-stage renal disease is characterized by a reduced Lp(a) catabolism.
Since liver is the almost only organ for Lp(a) biosynthesis, it is not astonishing that liver diseases are characterized by a gross reduction of plasma Lp(a). This was observed at first instance in patients with cholestasis, yet their Lp(a) reduction is transient if the patients are successfully treated. Other substances that are liver toxic including alcohol and several drugs have been also shown to significantly reduce Lp(a).
Related posts:
 Laboratory Assessment of Lipoproteins in Diabetes
Lipoprotein Subclasses and Cardiovascular Disease Risk in Insulin-Resistant Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Lipoproteins and Diabetic Nephropathy
The PPAR System in Diabetes
Laboratory Assessment of Lipoproteins in Diabetes
Lipoprotein Subclasses and Cardiovascular Disease Risk in Insulin-Resistant Diabetes
Emerging Lipoprotein-Related Therapeutics for Patients with Diabetes
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Lipoproteins and Diabetic Nephropathy
The PPAR System in Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


