Principles of radiation oncology
Philip P. Connell, MD  Ralph R. Weichselbaum, MD
Ralph R. Weichselbaum, MD
Overview
In recent decades, we have witnessed major technologic advances that have influenced how radiotherapy is delivered to cancer patients. Simultaneously, our understanding of the underlying basic biology that governs radiation effects in human cells and tissues has grown rapidly. Here, we summarize some of these key advances and place them into the context of the evolving paradigms that shape cancer therapeutics more generally. Key biologic topics covered in this chapter involve the molecular responses of cells to DNA damage by ionizing radiation, including the repair of DNA damage, cell cycle arrest and death, cellular signaling, and the influence of tumor–initiating cells (tumor stem cells) on post–treatment outcomes. As radiation generates an inflammatory microenvironment in tumors, drugs that modulate immune functions also represent a potential therapeutic opportunity. We also describe newer biology–based technologies that can accurately predict the intrinsic sensitivity of cells to radiation, and pharmacologic agents that can modulate this radiation sensitivity. Finally, this chapter reviews the major physics–based technologic innovations that have improved the accuracy and utility of clinical radiotherapy. Despite these advances, several unmet challenges continue to limit the impact of radiotherapy, and these remaining hurdles should be the factors that guide future innovations of our field.
Introduction
The medical specialty of radiation oncology began shortly after the discovery of X-rays and their effects on tissue in 1895.1 This first consisted of single large radiation exposures, using low-energy cathode ray tubes or radium-filled glass tubes positioned in close proximity to tumors. Technological developments between 1920 and 1950 improved the beam output and levels of deliverable energy. This was important because low energy (200–500 kV) X-rays used in these early periods had poor penetration into tissue, resulting in burned skin. The development of cobalt-60 units and linear accelerators in the 1950s to deliver “supervoltage” radiation energies (≥1 MeV) was a major advance, because these higher energy photons could penetrate more deeply into tissue. Another important advance was the use of fractionated therapy, in which treatment is divided into multiple small doses rather than one large dose. This enabled better tolerance of normal tissues to radiation. Additional improvements in treatment delivery technology, imaging, and cancer biology over subsequent decades have advanced the field of radiation oncology closer to the idealized goal of maximizing local cancer control while minimizing normal tissue toxicity.
General principles of radiotherapy in cancer treatment
Cancers tend to grow locally and spread systemically via lymphatic or hematogenous routes. Therefore, successful cure often requires therapies that are targeted toward all sites of involvement. The three major modalities of oncological therapy—surgery, radiotherapy, and chemotherapy—can be used alone or combined to address all sites. Surgery and radiotherapy are generally used to address local–regional areas of cancer risk. By its nature, surgery can be both therapeutic and diagnostic, as tumor removal provides tissue for histologic examination. Radiotherapy alone can sometimes offer a noninvasive alternative to surgery, with the possibility for organ preservation. In the adjuvant therapy setting, radiotherapy can be used before surgery to render tumor more resectable or to treat microscopic residual disease after surgery is completed. By contrast to these local therapies, chemotherapy can be used to address metastatic disease or to reduce the risk of potential micrometastasis. Chemotherapy can also act as a radiosensitizer, thereby increasing local tumor control provided by radiotherapy. The optimal use of these three modalities is tailored to the tumor type, anatomic location, stage of cancer, and other patient factors. Each modality carries a different set of risks that need to be balanced against one another to provide the optimal risk-benefit ratio for each individual patient.
Most clinical radiation regimens consist of fractionated therapy, in which treatment is divided into multiple small doses [expressed in units of Gray (Gy)] rather than one large dose. A common course of modern radiotherapy can average 6–8 weeks of treatment, with 5–6 daily treatments per week. The concept of fractionation is based on early empiric clinical observations that small daily radiation exposures can induce death in cancerous cells while allowing for normal tissue recovery. Radiobiologists have proposed various mechanistic explanations for this effect, but our understanding of this process continues to evolve. For example, the ability of normal tissue to repopulate itself with healthy cells represents one important component of normal tissue tolerance.
The impact of radiotherapy at the tissue level is commonly represented graphically by a sigmoid-shaped curve, as a function of increasing radiation dose on the x-axis (Figure 1). Ideally, the sigmoidal curve that describes tumor control probability is situated to the left of the curve that depicts normal tissue toxicity. The amount of separation between these two curves defines the therapeutic window, which describes the dose range wherein treatment may successfully eradicate tumor while maintaining normal tissue tolerance. These curves imply that for any given tissue, there exists a dose threshold above or below which incremental changes in dose yield little additional impact. However, within the steep portion of each curve, small changes in dose may yield large differences in terms of clinical impact. For any clinical situation there exists a unique relationship between the dose–response curves representing tumor kill and surrounding normal tissue toxicity, and these vary widely for different clinical circumstances. One should note that the curves in Figure 1 describe an idealized case, and in reality the relationship in sensitivity of actual tumors versus adjacent normal tissues is usually far more complex.
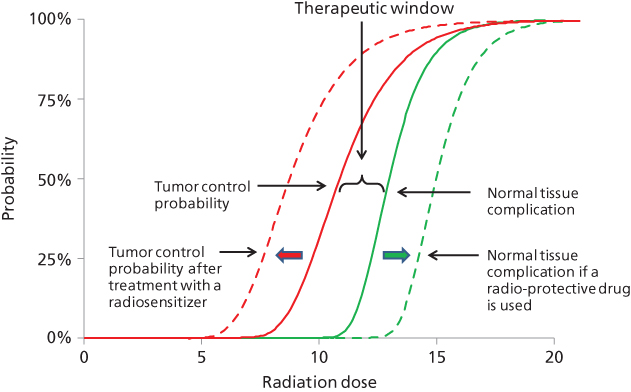
Figure 1 Effects of radiotherapy at the tissue level are graphically represented. The solid sigmoid-shaped curves denote idealized probability of tumor control and normal tissue complication risk. The dashed curves denote these same predicted effects following administration of drugs that modulate radiation sensitivity.
Biologic impact of radiation therapy
Radiation effects in cells and tissues
The molecular responses to radiotherapy in cells
One Gy leads to the formation of about 105 ionization events per cell, which yields approximately 1000–2000 single-stranded DNA breaks (SSBs) and about 40 double-stranded DNA breaks (DSBs) per nucleus. This DNA damage elicits the activation of a growing list of non-DNA repair/checkpoint-related mechanisms that mediate important cellular responses.2 Radiation-induced DSBs are generally thought to represent the principal lethal events occurring at doses relevant to clinical radiotherapy. This damage elicits numerous cellular responses including DNA damage recognition, transduction of the signal resulting in cell cycle checkpoint activation, induction and coordination of stress response genes, DNA repair, and/or activation of the apoptotic cascade (Figure 2). The MRN protein complex (Mre11, Rad 50, and Nbs1) and ataxia telangiectasia-mutated (ATM) protein are central sensors of DNA damage, and these proteins accumulate at DSBs rapidly. ATM, the related protein ataxia telangiectasia, and RAD3-related (ATR) are members of the PI-3 kinase family, which have central roles in recognizing DNA damage and initiating responses. ATM and ATR appear to perform partially overlapping functions; however, ATM preferentially recognizes DSBs, while ATR preferentially senses replication-blocking DNA lesions. They both phosphorylate downstream targets that modulate pathways that invoke cell cycle checkpoints, apoptosis, senescence, autophagy, and DNA repair. The chromatin protein histone H2AX is one such phosphorylation target, and its activation leads to chromatin structure becoming less condensed and receptive to the recruitment of repair proteins. Because H2AX phosphorylation is easily visible with fluorescent microscopic methods, it has become a commonly used marker of DSB formation and repair. Phosphorylated H2AX is centrally important to radiation response because it promotes the recruitment of other sensor/effector proteins including 53BP1, MDC1, and BRCA1.
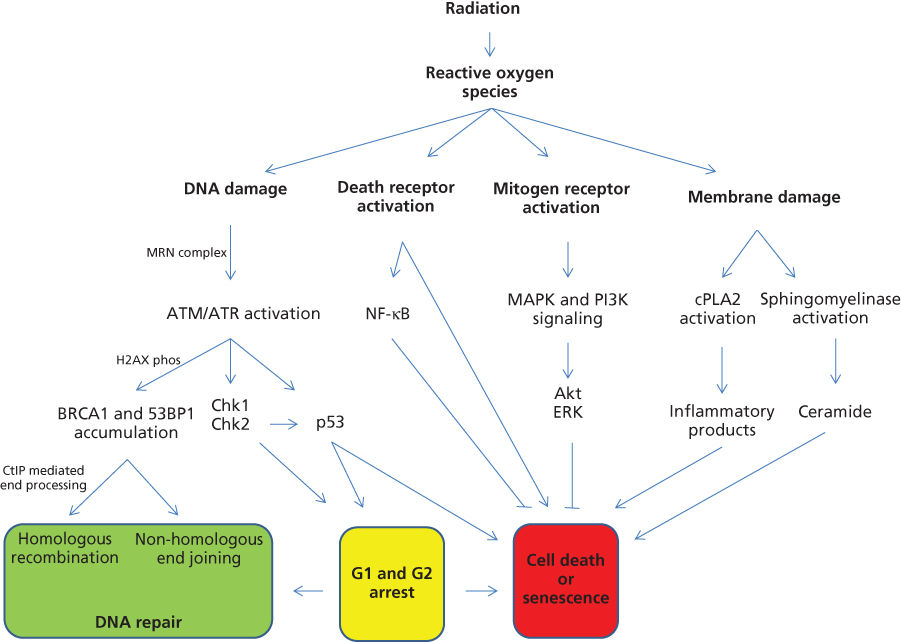
Figure 2 An overview is displayed for molecular responses that occur after radiation exposure.
Repair of DSBs in cells
Homologous recombination (HR) and nonhomologous end joining (NHEJ) are the dominant pathways that repair of DSBs. Both pathways act following the recruitment of the sensor/effector proteins, as previously reviewed.2 In HR, cells identify a stretch of homologous DNA and replicate the missing genetic information from this homologous DNA template.3 By comparison, the NHEJ pathway processes the broken DNA ends and religates them, sometimes using a region of microhomology.4 The NHEJ pathway is considered the dominant pathway for repairing radiation-induced DSBs during G0/G1. The generally accepted model involves the binding and processing of DNA ends by Ku70/80, Artemis, and DNA–PKcs complexes. The DSB is subsequently religated by the Ligase IV/XRCC4 complex.5 Because NHEJ sometimes requires processing of the DSB ends before religation, repair by this pathway has the potential to be error prone. HR, by comparison, is a generally error-free mode of DSB repair because the repair template is an undamaged sister chromatid. HR requires the action of many proteins including the key recombinase RAD51 and additional factors that promote its assembly on DNA, including BRCA2, RAD52, and the RAD51 paralogs.3 In addition to this DSB repair function, HR proteins promote tolerance of replication-blocking lesions (like damage by interstrand DNA cross-linkers) and collapsed replication forks. The ability of some common chemotherapeutic agents to act as radiosensitizers may result from their interference with these pathways.
Radiation-induced cell-cycle arrest and death
Cell-cycle arrest in G1 and G2 following radiation exposure was described by early pioneers like Hartwell, Nurse, and Hunt.2 Activated ATM and ATR lead to the phosphorylation of key downstream effector kinases such as Chk1 and Chk2. Following activation, Chk1 and Chk2 phosphorylate the phosphatase CDC25A, which in turn becomes degraded. As a result of this, CDC25A is unable to dephosphorylate and activate CDK2. The net result is cell-cycle arrest. Importantly, Chk2 also phosphorylates p53, leading to p21-mediated cell-cycle arrest. These damaged cells subsequently face a critical period, particularly if radiation damage is not completely repaired.6 Damaged cells may progress unrepaired into mitotic catastrophe. Alternatively, cells may simply lose proliferative capacity by undergoing senescence. Radiation-induced senescence is associated with the activation of the p16/RB and p53/p21 tumor suppressors. Alternatively, cells may undergo apoptotic death, which classically involves p53-dependent activation of the caspase cascade. Additionally, apoptotic death can occur via radiation-induced activation of cell surface “death receptors” (receptors of TNF, Fas, or TRAIL). Paradoxically, these death receptors can also generate cyto-protective effects in some situations by activating transcription factor nuclear factor kappa B (NFκB) to oppose apoptosis.
Major pathways involved in cellular signaling following radiation
Irradiation of tissue leads to the formation of radical oxygen species (ROS), which interact with many cellular contents including DNA. These ROSs, including superoxide and hydroxyl radicals, can deplete cellular stores of antioxidants such as glutathione,7 creating cellular stresses that stimulate a complex set of signaling cascades.8 For example, pathways normally used by cells to respond to mitogens are activated, which in turn promote survival, antiapoptotic responses, and transcriptional changes. The net effect can be variable and cell-type dependent, but this generally includes activation of cell surface receptors like the ErbB family that includes epidermal growth factor receptor (EGFR), which consequently activates other downstream pathways like the mitogen-activated protein kinase (MAPK) superfamily of cascades (ERK, JNK, and p38) and the phosphatidyl inositol 3 kinase (PI3K) pathways.2 These pathways tend to promote antiapoptotic signals via Akt and Erk signaling. Radiation can also trigger autocrine mechanisms, including the release of transforming growth factor alpha (TGFα), which binds and activates EGFR.8 Additional proinflammatory cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin-69 are also released, and these contribute to the bystander effect—a phenomenon where adjacent nonirradiated cells exhibit similar responses (described in detail in Dr. Grdina’s chapter on lonizing Radiation). Finally, radiation induces the breakdown of membrane-based sphingomyelin into ceramide, which is proapoptotic, independent of DNA damage.10 Similarly, radiation activates cytosolic phospholipase A2 (cPLA2), an enzyme that recognizes phospholipids on the cell membrane and degrades them into inflammatory products such as arachidonic acid and eventually eicosanoids. For example, lysophosphatidylcholine (LPC) is a product of cPLA2 that leads to activation of Akt and enhanced cell death.11
Tumor-initiating cells (tumor stem cells) in irradiated tumors and normal tissues
Despite some disagreements surrounding the nomenclature and basic biology of “tumor stem cells,” there clearly exists a population of tumor cells that exhibit an exclusive ability for self-renewal and differentiation into the heterogeneous lineages that promote tumor maintenance.12–14 These studies predict that targeting cancer stem cells may improve therapeutic outcomes. Interestingly, radiation of tumor cell populations is known to enrich cells expressing putative markers of “stemness,” suggesting that tumor stem cells are treatment resistant. For example, radiation leads to the enrichment of glial cancer stem cells that exhibit preferential activation of the DNA damage checkpoint responses and increased DNA repair capacity.15 The underlying mechanisms that mediate this radiation resistance remain unclear; however, aldehyde dehydrogenase 1 (ALDH1) may represent a partial explanation. High expression of ALDH1 protein in tumor cells confers a survival advantage, perhaps due to aldehyde-catabolizing activity. ALDH1 is also known to cooperate with Fanconi anemia-associated DNA repair genes. This suggests that ALDH1 activity might play a role in repairing adducts on DNA,16 thereby promoting radioresistance in stem cell clones. Another interesting feature of cancer stem cells is their propensity to repair DSBs using HR repair,17 suggesting that HR inhibitors might represent a therapeutic strategy for depleting cancer stem cell reservoirs in tumors. Additionally, salinomycin and thioridazine are compounds developed to target cancer stem cells18, 19; however, they have not yet been tested clinically with radiotherapy.
Development of radio sensitizing drugs
Tumors are frequently located immediately adjacent to radiosensitive normal tissues, which limits the level of radiation that can be safely administered. Therefore, a drug that could preferentially radiosensitize tumors would be very useful. In malignant gliomas, for example, lethality occurs as a direct consequence of local tumor recurrence after treatment: a tumor-selective sensitizer might increase tumor curability. By contrast, some tumors such as prostate cancers are already curable with radiotherapy; however, this requires very high doses, which carry significant toxicity. In such situations, a tumor-specific radiosensitizer might allow for dose de-escalation and reduction of side effects. Standard chemotherapeutic agents remain the most commonly used for increasing the local efficacy of radiotherapy. We recently published a review that details the progress of newer strategies of targeted radiosensitizers.2 An exhaustive discussion of these novel drugs is not possible in this chapter; however, various classes of agents are shown in Table 1. Many of these agents target proteins that sense radiation injury and coordinate the cellular responses, while others take advantage of the hypoxic or abnormal redox environments that exist in tumors. The ultimate success of any of these drugs will depend on their ability to improve the therapeutic index of radiation (see illustration of this concept in Figure 1). Unfortunately, the development of truly tumor-selective radiosensitizers has proved elusive.
Table 1 Drug candidates that may serve in modulating radiation effects
| Level (target) | Radiation sensitizer or protector |
| Reactive oxygen species | |
| Radicals | Amifostine, pyridoxamine, UROD RNAi |
| DNA damage response | |
| DSB recognition | Mirin, ATM/ATR inhibitors (e.g., KU-55933), DT01 |
| HR repair | RS-1, RI-1, modulators of RAD51 expression |
| NHEJ repair | DNA-PK inhibitors (e.g., NU7441, Salvicine) |
| PARP | Veliparib, olaparib |
| Chromatin organization | |
| HDAC | Vorinostat, belinostat, panobinostat |
| Cellular response | |
| Cell cycle arrest | Chk1/Chk2 inhibitors, CDK4/6 inhibitors |
| Mitogen signaling | Cetuximab |
| Pro-death signaling | Pifithrin, GSK-3β inhibitors, anticeramide, cytochrome c peroxidase inhibitors |
| Tumor microenvironment | |
| Inflammatory products | Statins |
| Hypoxia | Carbogen, efaproxiral, tirapazamine, MGd, EZN-2968 |
| Tumor stem cells | Salinomycin, thioridazine, HR inhibitors |
| Effects at tissue level | |
| Angiogenesis | Bevacizumab |