INTRODUCTION
SUMMARY
Primary myelofibrosis is one of several disorders in the spectrum of clonal myeloid diseases, malignant diseases that originate in the clonal expansion of a single hematopoietic multipotential cell reprogrammed by several somatic mutations. It is one of the eight neoplasms, including polycythemia vera and essential thrombocythemia, classified as a myeloproliferative disease by the World Health Organization. Approximately 90 percent of cases have a mutation in the Janus kinase 2 (JAK2) gene (~50 percent), the calreticulin (CALR) gene (~35 percent), or the thrombopoietin receptor (MPL) gene (~4 percent). The disease is characterized, classically, by anemia, mild neutrophilia, thrombocytosis, and splenomegaly. Occasional cases may present with bi- or tricytopenias (~15 percent). Immature myeloid and nucleated red cells, teardrop-shaped erythrocytes, and large platelets (megakaryocyte cytoplasmic fragments) are characteristic features of the blood film. The marrow contains an increased number of neoplastic dysmorphic megakaryocytes and increased reticulin fibers and, often later, collagen fibrosis. This reactive, polyclonal fibroplasia is the result of cytokines (e.g., transforming growth factor-β) released locally by the numerous neoplastic megakaryocytes. Osteosclerosis, also, may be present. The disease may be complicated by (1) portal hypertension and gastroesophageal varices as a result of a very large splenic blood flow and loss of compliance of hepatic vessels, (2) extramedullary fibrohematopoietic tumors that can develop in any tissue and lead to symptoms by compression of vital structures, and (3) abdominal vein thrombosis (Budd-Chiari syndrome). Newly developed JAK2 inhibitors are now first-line therapy for splenomegaly and constitutional symptoms (fever, night sweats, and weight loss). Other treatment has included hydroxyurea for thrombocytosis and massive splenomegaly, androgens, erythropoietin, or red cell transfusions for severe anemia, local irradiation of fibrohematopoietic tumors or of a massive, symptomatic spleen, or splenectomy for severe cytopenias, if splenic effects are not controlled by JAK2 inhibitors. Portosystemic shunt surgery may be required for gastroesophageal variceal bleeding. In younger patients, allogeneic hematopoietic stem cell transplantation can be curative and nonmyeloablative transplantation has been successful, at least up to age 60 years. The disease may remain indolent for years or may progress rapidly by further deterioration in hematopoiesis, by massive splenic enlargement and its sequelae, or by transformation to acute myelogenous leukemia.
Acronyms and Abbreviations
AML, acute myelogenous leukemia; bFGF, basic fibroblast growth factor; bp, base pair; CALR, calreticulin gene; CD, cluster of differentiation; CML, chronic myelogenous leukemia; FISH, fluorescence in situ hybridization; G6PD, glucose-6-phosphate dehydrogenase; G-CSF, granulocyte colony-stimulating factor; IL, interleukin; JAK2, Janus kinase 2 gene; MPL, thrombopoietin receptor gene; MRI, magnetic resonance imaging; PDGF, platelet-derived growth factor; TGF, transforming growth factor; TNFR, tumor necrosis factor receptor.
DEFINITION AND HISTORY
Primary myelofibrosis is a chronic clonal myeloid neoplasm characterized by (1) anemia; (2) neutrophilia and thrombocytosis or, in a minority, thrombocytopenia and leukopenia; (3) splenomegaly; (4) immature granulocytes, increased cluster of differentiation (CD) 34+ cells, erythroblasts, and teardrop-shaped red cells in the blood; (5) marrow fibrosis; and (6) osteosclerosis. The disorder originally was described by Heuck1 in 1879 under the title “Two Cases of Leukemia and Peculiar Blood and Bone Marrow Findings.” In his monograph, Silverstein traced the history of the concepts set forth during the first half of the 20th century to explain the pathogenesis of this disease, including its origin in the marrow, the appearance of extramedullary hematopoiesis, and the relationship of fibrosis to hematopoietic changes.2 The disease occurs, often, de novo but can develop as a later phase of polycythemia vera (Chap. 84), essential thrombocythemia (Chap. 85), or, rarely, chronic myelogenous leukemia (Chap. 89), or an atypical myeloproliferative disease (Chap. 89). It may be preceded by a prefibrotic phase (see “Special Clinical Features: Prefibrotic Primary Myelofibrosis” below). Numerous designations for the disease have been proposed or used, and different names were preferred in different countries.3 Primary myelofibrosis has been designated the most recent “official” name of the disease by a working group on classification of the World Health Organization.3 This compromise selection is unfortunate as the fibrosis is secondary, not primary, and the choice omits focusing on the central pathologic change: a clonal myeloid disease with the singular finding of neoplastic (dysmorphic) megakaryocytopoiesis.4 It is a disease of cells, not fibers, and is the only cancer in the medical lexicon not so designated; rather, it is named for an epiphenomenon in the extracellular matrix.
The discovery that the mutated Janus kinase 2 (JAK2) gene mutation can play a role in the causation and behavior of several myeloproliferative diseases5 and that approximately 50 percent of cases of primary myelofibrosis have a mutation of the JAK2 gene6 or, far less commonly, thrombopoietin receptor (MPL) gene has led to better understanding of the pathogenesis of the disease and its relationship to other myeloproliferative diseases. The JAK signaling pathways also represent an important new target for therapy. The subsequent observation of mutations of the calreticulin (CALR) gene in approximately 70 percent of patients without a JAK2 or MPL mutation has provided deeper insights into the pathogenesis of myelofibrosis, the opportunity to examine interactions among somatic mutations in the cells of patents, and the opportunity for new therapeutic approaches (see “Etiology and Pathogenesis” below).7
EPIDEMIOLOGY
Primary myelofibrosis characteristically occurs after age 50 years.2,8–16 The median age at diagnosis is approximately 65 to 70 years,8,12,13,14,17 but the disease also can occur in neonates and children.2,12,14,18,19,20 In infants, the disorder can mimic the classic disease or show certain features but not others, such as absence of hepatosplenomegaly.19 Familial infantile myelofibrosis mimics the adult disease and in some cases is transmitted by autosomal recessive inheritance.20,21,22,23 The occurrence of primary myelofibrosis in children usually is in the first 3 years of life.20,24,25 In young children, girls are afflicted with the disease twice as frequently as boys.19 In young and middle-age adults, the disease is similar to that in older subjects, although the proportion of indolent cases may be higher.18,20,26 In adults, the disease occurs with about equal frequency in men and women.8,12,13,14,15,16 Like virtually all clonal myeloid diseases,27 primary myelofibrosis can cluster in families, suggesting transmission of an unidentified predisposition gene.28,29,30 A large Swedish study found a significant relative risk (five- to sevenfold) for a familial occurrence of another myeloproliferative disease neoplasm, although not specifically primary myelofibrosis. The latter finding may relate to the small number of cases of primary myelofibrosis in that study.17 The incidence of the disease is approximately 0.5 cases per 100,000 population per year in northern European countries.31,32,33,34 A survey in Olmstead County, Minnesota reported an incidence of 1.5 case per 100,000 population per year and a median age of onset of 67 years,35 similar to other studies (see above in this section).
ETIOLOGY AND PATHOGENESIS
Exposure to high concentrations of benzene36,37,38 or very-high-dose ionizing radiation39 preceded the development of primary myelofibrosis in a very small number of cases in the past. Epidemiologic studies have not determined a relative risk of myelofibrosis after high-dose benzene exposure. Lower-level benzene exposure was not found to be associated with the risk of myeloproliferative disease in a comprehensive study.40 Thus, the few case reports must be viewed with caution in assessing causation. Benzene in exposures greater than 40 to 200 ppm-years is associated with an increased relative risk of acute myelogenous leukemia (AML)41,42 but not of chronic myelogenous leukemias (CMLs; Chap. 89) These levels of exposures are far above the current levels permitted by the Occupational Safety and Health Administration.
Reports of myelofibrosis in patients with lupus erythematosus, have suggested the possibility of immunologic-mediated hyperplasia of marrow connective tissue (see “Immune and Inflammatory Manifestations” below).2 These forms of myelofibrosis are different from the monoclonal multipotential hematopoietic cell disease, which is the principal type considered in this chapter.
The disease arises from the neoplastic transformation of a single hematopoietic multipotential cell, a conclusion derived from the presence of clonal cytogenetic abnormalities in patients with an identifiable chromosomal abnormality and in studies in women with primary myelofibrosis who also were heterozygous for isotypes A and B of glucose-6-phosphate dehydrogenase (G6PD).43,44 Although the nonhematopoietic tissues of these patients expressed both isotypes, each patient had blood cells with only one G6PD isotype. The findings strongly imply the blood cells of each patient arose from only one transformed stem cell. Furthermore, chromosome studies of colonies of hematopoietic progenitor cells in primary myelofibrosis established that the same clonal cytogenetic abnormality is present in erythroblasts, neutrophils, macrophages, basophils, and megakaryocytes.45 These studies were confirmed by (1) examining X-linked restriction fragment length polymorphisms in women with primary myelofibrosis with heterozygosity for X chromosome-linked genes46,47 and (2) verifying the presence of a mutation of codon 12 of the N-RAS gene in five blood cell lineages of a patient with the disease.48,49 Lymphocyte derivation from the clone has been noted using mutation in codon 12 of the RAS gene as the marker.48 Using fluorescence in situ hybridization (FISH) analysis, T and B lymphocytes were found to be derived from clonal expansion of a multipotential hematopoietic cell in three of four patients with primary myelofibrosis with a 13q– or 20q– clonal cytogenetic abnormality.50 Primary myelofibrosis can be distinguished from secondary myelofibrosis in women by clonality studies.51 The discovery of the JAK2V617F mutation has permitted this marker to be used in assessing clonality (as will mutations in CALR and MPL as their determination reaches the clinic). Mutated JAK2-containing cells were identified in all blood lineages of patients with primary myelofibrosis and in the common multipotential hematopoietic cell.52
The neoplastic hematopoietic stem cells in primary myelofibrosis containing the JAK2V617F mutation behave differently from the same cell population in polycythemia vera when studied in nonobese, diabetic, severe combined immunodeficient mice. Although studied in a nonhuman environment, the findings are consistent with the presence of JAK2V617F mutations in three phenotypically different myeloproliferative diseases: polycythemia vera, essential thrombocythemia, and primary myelofibrosis.53
Animal models allowed the derivation of the murine myeloproliferative leukemia (mpl) virus, carrying the oncogene v-mpl, which in mice produced a syndrome having features of a mixed idiopathic myelofibrotic–polycythemic disorder (Chap. 111).54 The availability of v-mpl led to the isolation of the thrombopoietin receptor and its ligand thrombopoietin.55 Later models of myelofibrosis and osteosclerosis, mimicking some of the important features of human primary myelofibrosis, were induced in mice by retroviral-mediated overexpression of thrombopoietin.56,57 The concomitant high levels of fibroblastic factors (transforming growth factor [TGF]-β1 and platelet-derived growth factor [PDGF]) resulted in intense fibrosis.58 In this model, increased osteoprotegerin was thought to be the principal cause of osteosclerosis.59 The disease was cured by murine hematopoietic stem cell transplantation.56 These findings were extended and led to the discovery of the human analogue of v-mpl, c-MPL that encodes the human thrombopoietin receptor. Two gain-of-function somatic mutations, MPLW515L and MPLW515K, were found to be associated with primary myelofibrosis and essential thrombocythemia.60,61 These gain-of-function mutations may also be germline and in such a case are associated with familial (hereditary) thrombocytosis.62
A syndrome in mice that results from the GATA-1 (low) mutation also leads to a phenotype that closely simulates human myelofibrosis. The mice gradually develop anemia, teardrop poikilocytes, myeloid immaturity, marrow fibrosis, extramedullary hematopoiesis, and overexpression of profibrotic cytokines in marrow.63 GATA-1 is a transcription factor required for normal megakaryocyte development. GATA-1 deficiency in mice leads to increased megakaryocytic proliferation, followed by myelofibrosis and osteosclerosis, as a result of exaggerated elaboration of fibroblast-inducing and osteoblast-stimulating factors.64,65
The discovery in 2005 that an activating somatic G→T point mutation (V617F) in JAK2 was associated with the three major myeloproliferative diseases—polycythemia vera, essential thrombocythemia, and primary myelofibrosis—has rapidly led to a fuller understanding of the pathogenesis of these diseases.48,66 A dominant, gain-of-function mutation in the gene JAK2 residing on chromosome 9p24, which encodes the JAK2 tyrosine kinase, is present in approximately 50 percent of patients with primary myelofibrosis, in approximately 95 percent of patients with polycythemia vera (Chap. 84), and in approximately 60 percent of patients with essential thrombocythemia (Chap. 85), but is absent in healthy individuals.67,68 In confirmation, the expression of the mutated human JAK2 gene transferred into mice can induce a myeloproliferative disease with features characteristic of the human disorders.69,70,71,72,73,74 Homozygosity results from allelic duplication as a result of uniparental disomy of chromosome 9p, not loss of the normal allele corresponding to the mutation.72
It is not yet precisely known how JAK2V617F, the most prevalent mutation, links the three diseases and what modifiers explain the dramatically different phenotype and expected survival of the patients with polycythemia and primary myelofibrosis. At least four other modifying factors have been proposed to account for the different phenotypes and the apparent absence of the mutation in a high proportion of patients with primary myelofibrosis: (1) gene dosage, (2) germline modifiers, (3) predisposition alleles, and (4) additional somatic mutations.66,68,73 An example of each influence follows. There is an increasing JAK2V617F allele burden from essential thrombocythemia, to polycythemia vera, to primary myelofibrosis.74 For example, the JAK2V617F allele burden may be a key determinant of the degree of myeloproliferation and myeloid metaplasia reflected by significantly higher levels of white blood cell counts, CD34+ cell counts, lower platelet counts, and a higher frequency of splenomegaly in homozygous polycythemia vera patients compared to their heterozygous counterparts. These findings are consistent with JAK2V617F-positive chronic myeloproliferative disorders as a biologic continuum with phenotypic presentation in part influenced by JAK2V617F mutational load.74 Myeloproliferative neoplasm predisposition alleles could provide a selective advantage for the development of mutations in the JAK2 signaling pathway. A number of pre-JAK2 alleles, which arise in cells prior to JAK2 mutation, may contribute to the phenotype displayed.68
A mutation in the thrombopoietin receptor gene, MPL, (MPL515L/K) has been found in some mutant JAK2-negative patients with primary myelofibrosis. The finding of an activating JAK2 (~50 percent of patients) or MPL (~4 percent of patients) mutation, which employs JAK2 for signaling, reinforces the critical role of unregulated activation of the JAK-STAT (signal transducer and activator of transcription) signaling pathway in the pathogenesis of primary myelofibrosis.75,76,77 Using next-generation gene whole-exome sequencing, primary myelofibrosis patients who did not have mutated JAK2 or MPL genes (approximately half of all patients) were only found to have an occasional somatic mutation.78
It came as a surprise that nearly a decade after the description of the JAK2 mutation in primary myelofibrosis and related myeloproliferative diseases, two groups reported, simultaneously, the presence of CALR mutations in approximately 70 percent of patients with primary myelofibrosis who did not carry the JAK2 or MPL mutations (35 percent of total population).79,80 The CALR mutations were either from deletions or insertions, explaining the failure to identify them by Sanger sequencing technology. The 52-bp deletion (type 1 mutation) or 5-bp insertion (type 2 mutation) are the most frequent types, accounting for approximately 85 percent of the CALR mutations. The CALR mutations are missense for the gene’s final domain, encoding the calcium binding site.
Patients with primary myelofibrosis and JAK2, CALR, or MPL mutations may have an additional two to four somatic mutations. Some of the principal genes mutated are TET2, ASXL1, DNMT3A, EZH2, and IDH1, which are implicated in epigenetic regulation, and TP53 and CBL.7 As many as five somatic mutations could be identified in 3 percent of patients, whereas 12 percent of patients with primary myelofibrosis did not have an identifiable somatically mutated gene, using targeted next-generation whole-exome gene sequencing.7 However, the proportion of these additional somatic mutations was less that that detected by similar studies in polycythemia vera,81 suggesting that these sequencing technologies, capable of uncovering only approximately 1 percent of the genome, may leave unidentified additional somatic and germline mutations present in a much greater proportion of primary myelofibrosis patients.
Isolated genetic findings in individual patients have included (13q14) deletions, mutation or overexpression of the retinoblastoma gene,82,83 NF1 (17q11) deletions,84 RAS mutations in approximately one in 20 patients studied, and occasional patients with mutations in KIT.71 Uniparental disomy has been found on chromosomes 9p (site of JAK2, creating a second allele of V617F) and 1p.83
HMGA2, a gene on chromosome 12, normally is not expressed in humans and is implicated in mesenchymal tumors. HMGA2 was expressed in 12 of 12 patients with idiopathic myelofibrosis studied, implying that, if confirmed, expression of this gene in myeloid cells may play a role in the disease.87
Neoplastic megakaryopoiesis is the most prominent alteration in this clonal myeloid disease and is responsible for most of its major manifestations. Constitutive mobilization and circulation of CD34+ cells are prominent features of the clonal expansion. This phenomenon, probably, is the result of epigenetic methylation of the CXCR4 promotor, a resultant decrease in CXCR4 mRNA, decreased expression of CXCR4 on CD34+ cells, and their resultant enhanced migration into the blood in primary myelofibrosis patients.85
Circulating CD34+ cells in patients with primary myelofibrosis generate about 24-fold the number of megakaryocytes in culture than do CD34+ cells from normal subjects, express increased levels of BCL-XL, and have delayed apoptosis.86,87 Media conditioned with CD61+ cells (presumptive megakaryocytes) elaborated greater quantities of growth factors and proteases, including TGF-β and metalloprotease-9, than did CD61+ cells generated from normal CD34+ cells.
Circulating CD34+ cells in patients with primary myelofibrosis also had a higher expression of eight genes (CD9, GAS2, DLK1, CDH1, WT1, NFE2, HMGA2, and CXCR4) than did normal CD34+ cells. These genes or subsets of them are likely related to disease pathogenesis and were shown to be related to specific manifestations in patients (e.g., increased expression of CD9 and DLK1 with platelet count and WT1 with severity score).88
Microvessel density and marrow blood flow are increased in patients with myelofibrosis. These changes may be related to an increase in circulating endothelial cell progenitors.89 The endothelial cells examined by laser microdissection, cell sorting, or cell culture from spleen capillaries and splenic vein contained the JAK2V617F mutation in 12 of 18 patients studied who had the mutation in their granulocytes. A definitive explanation for this finding has not been established.90
Neoplastic myeloproliferation usually is the dominant marrow abnormality in the granulocytic and megakaryocytic lineages resulting in intensely cellular marrows and mild to moderate blood granulocytosis and thrombocytosis. Hypocellular hematopoiesis, resulting from exaggerated apoptosis of very early precursors, can be present initially or emerge later, leading to granulocytopenia and/or thrombocytopenia. Anemia is a frequent finding and results from a combination of decreased erythropoiesis, shortened red cell survival, and the effects of splenomegaly on the distribution of red cells in the circulation. Hemolysis can be a prominent factor in some cases. Neoplastic megakaryocyte expansion and intense dysmorphogenesis of megakaryocytes are constant features of the disease. Even in intensely fibrotic marrows with severe decreases in erythroid and granulocytic precursors, clusters of megakaryocytes are easily found interspersed between collagen bundles. The term megakaryocytic myelosis, one of the many synonymous terms for the disease, exemplifies the constancy of this finding. The dominance of megakaryopoiesis may relate to the average fivefold overexpression of FKBP51 in megakaryocytes in primary myelofibrosis and the marked predisposition of CD34+ cells to differentiate into megakaryocytes (see “Centrality of CD34+ Cell Egress and Neoplastic Megakaryocytopoiesis” above). FKBP51 increases resistance to apoptosis, possibly by an effect through the calcineurin pathway.91 Thus, the designation “chronic megakaryocytic leukemia” would be a more accurate designation for primary myelofibrosis, if one used an internally consistent classification.7 Although elevated levels of thrombopoietin (and interleukin [IL]-6 and IL-11) are found in the serum of patients with primary myelofibrosis, their etiologic role in the human disease is unresolved.92 A marked increase in the thrombopoietin receptor MPL is observed on the platelets and megakaryocytes of a proportion of patients with primary myelofibrosis.93 Despite the animal models of thrombopoietin-induced myeloproliferation and osteomyelofibrosis and the apparent abnormality of MPL receptor sites on human megakaryocytes, autonomous megakaryocyte growth, characteristic of human primary myelofibrosis marrow in culture, has not been associated with either an autocrine effect of MPL ligand (thrombopoietin) or of a mutation in MPL.
Four of the five major types of collagen94 are present in normal marrow: type I in bone, type III in blood vessels, and types IV and V in basement membranes. The fine reticulin fibers that appear after silver impregnation of marrow are principally type III collagen. They do not stain with trichrome dyes. The thicker collagen fibers are principally type I collagen and stain with trichrome dyes, but do not impregnate with silver. The amount of the very fine fibrous network barely perceptible in normal marrow that is stained by silver impregnation techniques95 increases in the marrow of patients with primary myelofibrosis (Table 86–1 and Fig. 86-1C).96 The fibrous network contains collagen and occasionally progresses to include thick collagen bands that are evident with trichrome stains. Collagen types I, III, IV, and V are increased in myelofibrosis, but type III collagen is increased uniformly and preferentially.97,98,99,100,101,102,103,104 The latter occurrence accounts for the increased plasma concentration of procollagen III aminoterminal peptide, a component of collagen type III, which is cleaved during collagen biosynthesis.96,101,102 Serum prolyl-hydroxylase and marrow and plasma fibronectin also increase in patients with idiopathic myelofibrosis or myelofibrosis from other causes.98,99 Several other matrix materials are increased in marrow or plasma (Tables 86–1 and 86–2).105–119
I. Marrow stroma A. Increased amount of 1. Total collagen (hydroxyproline)97,101 2. Type I collagen97,98,99,103 3. Type III collagen97,98,99,103 4. Type III procollagen98,101,103,104 6. Matrix metalloproteinase-14107 7. Bone morphogenetic protein108 10. Tenascin112 11. Vitronectin113 12. Microenvironment transforming growth factor-β,114 basic fibroblast growth factor,114 and substance P115 B. Decreased amount of 1. Collagenase107 II. Plasma A. Increased concentration of 1. Prolylhydroxylase116 2. C-terminal peptide of procollagen type I100 3. N-terminal peptide of procollagen type III99,101,117,118 7. Hyaluronan119 |
| PREFIBROTIC STAGE |
Anemia may be absent or mild Leukocytosis may be absent or slight Thrombocythemia is invariable BCR-ABL fusion gene absent Presence of JAK2, CALR, or MPL mutations indicative of diagnosis of myeloproliferative disease (one of these mutations present in ~90% of patients) Cellular marrow with mild increase in granulopoiesis; increased megakaryocytes, clusters of very dysmorphic megakaryocytes and megakaryocytic nuclei; no to very slight increase in reticular fibers on silver stain Palpable splenomegaly infrequent Absent or slight anisopoikilocytosis including teardrop red cells |
| FULLY DEVELOPED STAGE |
Marrow reticulin fibrosis plus or minus collagen fibrosis BCR-ABL fusion gene absent JAK2, CALR, or MPL mutation in approximately 90% of patients Splenomegaly Anisopoikilocytosis with teardrop red cells in virtually every oil immersion field Immature myeloid cells in blood Increased CD34+ cells in blood Nucleated red cells in blood Marrow usually hypercellular but invariably has increased megakaryocytes, clusters of highly dysmorphic megakaryocytes, and megakaryocyte bare nuclei regardless of overall marrow cellularity |
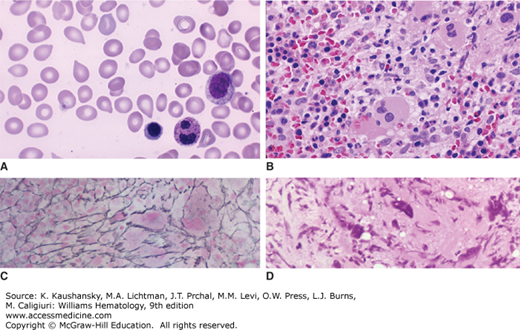
Figure 86–1.
Blood film and marrow sections from patients with primary myelofibrosis. A. Blood film. Characteristic teardrop poikilocytes, a nucleated red cell, and a segmented neutrophil with a dysmorphic nucleus are evident. B. Marrow section. Low power. Hypercellular marrow with increased number of hypolobular megakaryocytes. C. Marrow section. Silver impregnation stain. Marked increase in argentophilic fibers representing collagen type III (reticulin). D. Marrow section. Collagen fibrosis with extensive replacement of marrow with swirls of collagen fibers. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Marrow fibrosis in primary myelofibrosis is most closely correlated with increased neoplastic and dysmorphic megakaryocytes in the marrow. Even densely fibrotic marrow with little residual granulopoiesis or erythropoiesis usually has numerous megakaryocytes scattered throughout the fibrotic areas.96,120 The increased pathologic emperipolesis (the entry of neutrophils and other marrow cells into the canalicular system of megakaryocytes), evident in human primary myelofibrosis and in mouse models, suggests this may be an additional mechanism of α-granule injury and release of TGF-β and PDGF.121 Animal models also indicate that marrow monocytes and macrophages may play a subsidiary role in the induction of fibrosis.121,122,123 Secretion of PDGF, basic fibroblast growth factor (bFGF), and TGF-β from monocytes that are part of the clone have the potential to act as myeloproliferative growth factors and profibrotic cytokines.114
The increased content of marrow collagen types I and III results from release of fibroblast growth factors, which include PDGF,123,124 epidermal growth factor,126 endothelial cell growth factor,126 TGF-β,114,127,128 and bFGF,114,129 each of which is present in megakaryocyte α granules. Other factors, such as tumor necrosis factor α, IL-1α, IL-1β, and lysyl oxidase, which can be released from marrow cells, also can stimulate fibroblasts.130,131,132 Platelet factor 4, also derived from megakaryocytes, inhibits collagenase and could contribute to collagen accumulation,120 although studies showing a poor correlation between plasma platelet factor 4 concentration and marrow fibrosis have dampened enthusiasm for the role of this factor.133 Substance P, a peptide that acts as a neurotransmitter and a modulator of immune and hematopoietic functions, is increased in the fibrotic marrow and colocalizes with fibronectin. It is angiogenic and is a fibroblast mitogen.115 Its precise role in the complex interactions among fibroblasts, cytokines, and matrix protein deposition is not clear. The high urinary excretion of platelet-derived calmodulin, a putative fibroblast growth factor, in patients with myelofibrosis has added this compound to the array of factors that may contribute to the fibroplasia.130 The plasma level of matrix metalloprotein III is decreased and the level of tissue inhibitor of metalloproteinase is increased in patients with idiopathic myelofibrosis.134 The expression of matrix metalloproteinase-14 in marrow increases by nearly two orders of magnitude as fibroplasia progresses during the course of the disease; and, megakaryocytes and endothelial cells are the major sources of this protein.107 Neutrophil collagenase (matrix metalloproteinase-8) content is decreased early in the disease.107 Bone morphogenetic proteins (BMPs) also are implicated as a contributory factor in fibroplasia. BMP1, BMP6, BMP7, and BMP-receptor 2 are increased in marrow in myelofibrosis as a result of release from megakaryocytes and stromal cells. These proteins are activators of latent TGF-β1 and processors of collagen precursors. In addition, TGF-β1 induces release of BMP6.108
This complex combination of alterations contributes to matrix deposition. The pathogenetic role of released growth factors in fibroplasia is not completely understood. Generalizations from in vitro experiments or correlation between two variables provide only a limited perspective. For example, TGF-β can stimulate or inhibit fibroblast growth, depending on the repertoire of other growth factors in the environment.127,128
Fibroplasia is associated with an increase in the number and size of marrow sinuses,111 the number of endothelial cells,135 an increase in vascular volume in the marrow,106 and an increase in blood flow through the marrow.136,137 These processes are responsible for the increase in marrow collagen types IV and V and laminin synthesized by endothelial cells in the marrow of patients.126
The fibroblastic proliferation in marrow is not an intrinsic part of the abnormal clonal expansion of hematopoiesis.138 In cases of primary myelofibrosis in which G6PD isoenzyme studies or chromosome karyotyping establish monoclonal growth of hematopoietic cells, marrow fibroblasts contain both G6PD isoenzymes and do not share the clonal chromosome abnormality.139 The findings strongly imply that the fibroblasts differentiate from a primordial cell different from the neoplastic hematopoietic stem cell in primary myelofibrosis and that fibroblast proliferation and enhanced collagen synthesis are secondary results of abnormal hematopoiesis.
Extramedullary hemopoiesis is almost always present in liver and spleen, where it contributes to organ enlargement.8,9,10 Escape of progenitor cells from marrow and their lodgment in other organs contributes to extramedullary blood cell formation. Reversion of the liver and spleen to their fetal hematopoietic functions (metaplasia) is not a major factor in extramedullary hematopoiesis, and quantitatively significant, effective hematopoiesis does not occur outside of the marrow (see also “Extramedullary (Fibrohematopoietic) Tumors” below).
CLINICAL FEATURES
Some patients are asymptomatic at the time of diagnosis; in which case the disease is detected by medical examination for an unrelated reason. In symptomatic patients, fatigue, weakness, shortness of breath, pruritus, and palpitations are nonspecific but frequent complaints.9,10,11,12,13 Fatigue is the most frequent self-reported complaint and is disproportionate to the degree of anemia. Weight loss is common, but anorexia is less so, and fever and night sweats may occur. The term constitutional symptoms, often used in studies of the response to treatment of myelofibrosis, refers specifically to the aggregate occurrence of fever, weight loss, and night sweats.140 A dragging sensation in the left upper abdomen caused by an enlarged spleen or early satiety from encroachment of the spleen on the stomach may occur. Severe left upper quadrant or left shoulder pain can occur from splenic infarction and perisplenitis. Patients may report unexpected bleeding. Occasionally, bone pain is prominent, especially in the lower extremities. Fever, weight loss, cachexia, night sweats, and bone pain are more frequent later in the course of the disease and are related to the increase in circulating inflammatory cytokines that are a key feature of the disease (see “Immune and Inflammatory Manifestations” below).
Hepatomegaly is detectable in two-thirds of patients, and splenomegaly is present on palpation or imaging studies in almost all patients at the time of diagnosis.8,9,10,11,12 The spleen is mildly enlarged in one-fourth, moderately enlarged in half, and massively enlarged in approximately one-fourth of patients. Muscle wasting, peripheral edema, and purpura are present infrequently. Bone tenderness may be present. The latter signs may develop in a larger proportion of patients over the course of the disease.
Neutrophilic dermatosis, a syndrome that closely mimics the raised and tender plaques of Sweet syndrome, may occur.141,142,143 It can be the presenting or a significant complicating feature, and can progress to bullae or pyoderma gangrenosum.141,144 The dermatopathology of neutrophilic dermatosis is different from leukemia cutis and is unrelated to infection or vasculitis. The predominant histologic lesion is an intense polymorphonuclear neutrophilic infiltrate.
Skin infiltrates related to hematopoietic cells (leukemia cutis) are uncommon.145 These cutaneous lesions may have myeloid cells with giant cells carrying CD61 markers characteristic of megakaryocytes.146,147 Skin lesions representing cutaneous fibrohematopoietic tumors may occur.
The presenting findings of the clonal myeloid diseases are changing because of more and earlier access to healthcare in industrialized countries (see Table 86–2). A subset of patients, perhaps as many as 25 percent, with primary myelofibrosis present without overt reticulin fibrosis in the marrow.148,149 Blood hemoglobin may be normal and white cell count mildly elevated. The classic findings of frequent teardrop red cells, myelocytes, and nucleated red cells in the blood film and palpable splenomegaly often are absent. Thrombocytosis is a constant finding. Essential thrombocythemia is closely simulated, but observation eventually shows evolution to primary myelofibrosis. The most important distinction with essential thrombocythemia is the nature of the megakaryocytic expansion.150 In primary myelofibrosis, bizarre changes are evident with wide variation in megakaryocyte size, from very small to giant size cells. Nuclear lobulation is abnormal, with bulky multilobulation, hypolobulation, and free megakaryocyte nuclei in the marrow spaces. In essential thrombocythemia, megakaryocytes are increased but they do not display the dysmorphia observed in myelofibrosis. The prefibrotic disease usually evolves into fully developed myelofibrosis over a period of years. Investigators, evaluating histopathology in a blind fashion, have confirmed the entity of prefibrotic myelofibrosis and this abnormality predicts for progression to an overt primary myelofibrosis and, thus, has an impact on the risk of progression to acute leukemia and prognosis in general.151
The appearance of symptoms or signs leading to (1) identification of a mass on imaging regardless of location, (2) appearance of signs or symptoms of an effusion in the thorax or abdomen, (3) unexpected neurologic signs, or (4) another finding that appears unexpected in a patient with primary myelofibrosis should be considered a fibrohematopoietic (extramedullary) tumor(s) until proven otherwise. Foci of hematopoiesis may become clinically apparent as fibrohematopoietic tumors in the adrenal glands,152,153 renal parenchyma,154,155,156 and lymph nodes.157,158,159 Tumors composed of hematopoietic tissue, sometimes with intense fibrosis, can develop in the bowel,160,161,162,163 breast,164,165,166 liver,167,168 lungs,169,170,171 mediastinum,172 pleura and mesentery,169,171,173 skin,174,175 synovium,176 thymus,169 thyroid,177 thorax,178 prostate,179 spleen,180 or urinary tract.178,181,182,183,184
Extramedullary hematopoiesis in the intracranial or intraspinal epidural space can lead to serious neurologic complications, including subdural hemorrhage,185 delirium,185,186 increased intracranial pressure,187 orbital apex syndrome,188 papilledema,189 cerebral tumor,190 coma,191 motor and sensory impairment,192,193 spinal cord compression,194,195 and limb paralysis.195,196 Intraspinal myelography,193,194,195,196,197,198,199 computed axial tomography,185,187,191,192,193,194,195,196,197,199 positron emission tomography after 52Fe infusion,186 and magnetic resonance imaging (MRI)198,199 each has been used to define the location and nature of the masses.
Hematopoietic foci on serosal surfaces can produce effusions, sometimes massive, in the thorax,178,180 abdomen,172,173,201,202 and pericardial space.203,204,205,206 The effusion fluid often contains megakaryocytes, immature granulocytes, and, occasionally, erythroblasts.207,208,209 Splenectomy is sometimes followed by extramedullary hematopoietic tumors in soft tissues,210 in body cavities, or on serosal surfaces,209 perhaps as a result of an increase in circulating hematopoietic progenitors211 and loss of the filtration function of the spleen. In rare cases, extramedullary soft-tissue megakaryoblastic tumors simulate the myeloid sarcoma (synonyms: chloroma, granulocytic sarcoma) of other types of myelogenous leukemia.212,213
In patients with primary myelofibrosis, there can be a massive increase in splenoportal blood flow and a decrease in hepatic vascular compliance or the presence of hepatic vein thrombosis, either of which can result in severe portal hypertension, ascites, esophageal and gastric varices, intraluminal gastrointestinal bleeding, and hepatic encephalopathy.214,215,216 The hepatic venous pressure gradient, normally less than 6 torr, is markedly elevated.217
Perisinusoidal fibrosis,218,219,220 collagen bundles in the spaces of Disse,219 perisinusoidal fibroplasia,218,219,220,221 and foci of hematopoietic cells219,222 appear to contribute to the decreased sinusoidal compliance. Portal vein thrombosis is a complication of primary myelofibrosis and occasionally precedes disease onset.223
Rarely, portal hypertension is accompanied by pulmonary hypertension and may result from pulmonary fibrosis171 or hydrodynamic factors.224 Pulmonary arterial hypertension, also, may be the principal problem.225,226 Although as many as one-third of patients with primary myelofibrosis have an elevated systolic pulmonary artery pressures (>35 torr), the fraction that is symptomatic is very small. Elevated vascular endothelial growth factor (VEGF) levels, elevated circulating endothelial cells, and elevated marrow microvessel density in patients suggest that proangiogenic factors may contribute to the hypertension.227 Contrariwise, secondary myelofibrosis with polyclonal hematopoiesis and normal blood CD34 cell concentrations frequently occurs in patients with primary pulmonary hypertension.228
Abnormalities of humoral immune mechanisms have been observed in up to half of patients with primary myelofibrosis.229,230,231,232,233,234 The array of immune products and events reported includes anti–red cell antibodies,233,234,235,236,237 antiplatelet antibodies,238,239 antinuclear antibodies,229,230,234 elevated plasma-soluble IL-2 receptor,240 anti-Gal (galactoside determinants) antibodies,241 anti–γ-globulins,229,230,234 antiphospholipid antibodies,234,242 antitissue or organ-specific antibodies,231,233 and circulating immune complexes,234,243,244,245 as well as complement activation,234,246 immune complex deposition,231 interstitial immunoglobulin deposition,231 increased numbers of marrow plasmacytoid lymphocytes,231,243 and development of amyloidosis.244,245,246,247
Inflammatory cytokines, including IL-1β, IL-6, IL-8, tumor necrosis factor (TNF)-α, TNF receptor II (TNFRII), and C-reactive protein also are markedly elevated and play a role in the constitutional symptoms seen in patients with progressive disease,248 which explains the often rapid, symptomatic improvement in patients treated with JAK2 inhibitors before splenic size is reduced.
Occasional reports of nonclonal secondary myelofibrosis associated with lupus erythematosus,249,250,251,252,253,254 vasculitis,255 polyarteritis nodosa,234,255 ulcerative colitis,256 scleroderma,257 biliary cirrhosis,237,258,259 Sjögren syndrome,260 and acute reversible myelofibrosis responsive to glucocorticoids,261 although fundamentally different processes from primary myelofibrosis, have raised the possibility that immune mechanisms play a role in the development of marrow fibrosis in some circumstances.
A large proportion of patients have osteosclerosis at diagnosis or develop osteosclerosis during the course of the disease,11,12,13,14,15,262,263,264,265 as reflected by increased bone density on imaging studies and histomorphometric analysis of a bone biopsy (Table 86–3).263,264,265,266,267,268 The proximal femur and humerus, pelvis, vertebrae, ribs, and skull may be involved. MRI can uncover evidence of new bone formation and periosteal thickening. Lumbar spine dual-energy x-ray absorption studies and quantitative computed tomography provide evidence for increased bone formation, bone thickening, and higher proportions of cancellous and of woven bone.268,269 Osteolytic lesions are rare270 and may reflect a myeloid sarcoma.271 Periostitis, although infrequent, can lead to debilitating bone pain.272
|
The risk of arterial and venous thrombosis is elevated in patients with primary myelofibrosis, although not to the degree seen in polycythemia vera or essential thrombocythemia.273 Approximately 10 percent of patients with myelofibrosis will develop a significant thrombotic event during the first 4 years of the disease. The two principal risk factors are an elevated leukocyte count and age, but not platelet count.274 In a large multicenter study of 707 patients with primary myelofibrosis, thromboses occurred in 7.2 percent of patients over the period of observation, or 1.8 percent patient-years. The combination of the JAK2 mutation, leukocytosis, and age predicted the highest incidence of thrombosis.275 The cardiovascular risk factor, such as hypertension, hypercholesterolemia, or smoking, further increase thrombotic risk. Multiple thrombotic episodes may occur; and, the thrombotic event may occur at or just before diagnosis.
Noncirrhotic splanchnic vein thrombosis includes hepatic vein thrombosis (Budd-Chiari syndrome) and portal vein thrombosis, which may occur with minimal evidence of a clonal myeloproliferative disease. In the past marrow examination or evidence of erythropoietin-independent colony growth was used to determine if an occult or incipient myeloproliferative disease may underlie the thrombosis. Now JAK2 mutational analysis can be done and is positive in 35 percent of seemingly idiopathic hepatic vein thrombosis and 25 percent seemingly idiopathic portal vein thrombosis.276 Presumably, future use of CALR gene mutational analysis will increase the proportion of cases of hepatic vein thrombosis indicative of an underlying occult myeloproliferative disease.
LABORATORY FEATURES
The range of values for blood cell counts at the time of diagnosis is very broad. Normocytic–normochromic anemia is present in most, but not all, patients (see Table 86–2).8,9,10,273,274,275,276,277,278,279,280 Mean hemoglobin concentration in a series of patients at diagnosis was approximately 9 to 12 g/dL (range: 4–20 g/dL).8,9,10,11,12,13,14,15,16,279,280 Anisocytosis and poikilocytosis are a constant finding. In all cases, teardrop-shaped red cells (dacryocytes) are present in sufficient number to be found in every oil immersion field (see Fig. 86–1). Nucleated red cells are present in the blood film of most patients and average 2 percent of nucleated cells (range: 0 to 30 percent). The percentage of reticulocytes is mildly increased but may vary widely in a given case. A decreased blood hemoglobin may be attributed in part to the expansion of plasma volume and a higher than normal proportion of the red cell volume in an enlarged spleen. Ineffective erythropoiesis can result in a decrease in red cell mass.277 Erythroid hypoplasia is present in many patients.281,282 In some patients, hemolysis may be prominent, and polychromatophilia and very elevated reticulocyte counts can occur.278,279 The antiglobulin (Coombs) test usually is negative, but red cell autoantibodies can develop and lead to immune-mediated hemolysis,234,235,236,283 which rarely has been a presenting finding of the disease.236 Occasional patients have had a positive acid hemolysis and sucrose hemolysis test, reflecting a concurrent clone of cells consistent with paroxysmal nocturnal hemoglobinuria.284 Acquired hemoglobin H disease, coincident with typical white cells and platelet changes of myelofibrosis, can occur285 and results in hemolysis, hypochromic–microcytic red cells, marked poikilocytosis, and hemoglobin H inclusions that stain with brilliant cresyl blue. Red cell aplasia, in association with primary myelofibrosis, has been observed.280,286
The total white cell count usually is usually moderately elevated as a result of granulocytosis.8,9,10,11,12,13,14,15,16 The mean total blood white cell count was 10,000 to 14,000/μL (10 to 14 × 109/L) in four large studies. Neutropenia, however, is present in approximately 20 percent of patients at the time of diagnosis.8,9,10,11,12,13,14,15,16 The range of white cell counts was 400 to 237,000/μL (0.4 to 237.0 × 109/L) at the time of diagnosis.8,9,10,11,12,13,14,15,278,279 Myelocytes and promyelocytes are present in small proportions in the blood film in most patients, and a low proportion of blast cells (0.5 to 2.0 percent) may be found in the blood film. The blood blast cells range from 0 to 20 percent at the time of diagnosis. In patients with blast counts at the high end, which is unusual at presentation, the disease merges with or may progress rapidly to AML. Hypersegmentation, hyposegmentation (acquired Pelger-Huët anomaly), and abnormal granulation of neutrophils may be present.8,9,10,11,12,13,14,15,16 Neutrophil alkaline phosphatase scores may be elevated (25 percent of patients) or decreased (25 percent of patients).287 The percentage of basophils may be slightly increased.279 The mean platelet count in patient series ranges from 175,000 to 580,000/μL (175 to 580 × 109/L) at the time of diagnosis. Individual platelet counts can range from 15,000 to 3,215,000/μL (15 to 3215 × 109/L).8,9,10,11,12,13,14,15,16,278,279 The platelet count is elevated above the normal upper limit in approximately 40 percent of patients.279 Mild to moderate thrombocytopenia is present in approximately one-third of patients at the time of diagnosis, particularly if splenomegaly is massive. Giant platelets and abnormal platelet granulation in the blood film are characteristic features of the disease.
Approximately 10 percent of patients present with pancytopenia because of severe impairment of hematopoiesis affecting each cell lineage, coupled with sequestration in a massively enlarged spleen. Pancytopenia usually is associated with intense marrow fibrosis.
Increased concentrations of multipotential,288,289 granulocytic,290,291 monocytic,291 erythroid,292 and megakaryocytic293 progenitor cells are present in the blood of patients, as measured by clonogenic assays in semisolid cultures. The frequency of hematopoietic progenitor cells in the blood is correlated with the extent of marrow reticular fiber density.293 Megakaryocytes also are present in the systemic venous blood.294 An increase in blood CD34+ cells is very characteristic of primary myelofibrosis, and the concentration of these cells lends weight to the diagnosis. The height of the CD34+ cell count is correlated with the extent of disease and disease progression. Greater than 15 × 106/L blood CD34+ cells is virtually diagnostic of primary myelofibrosis, and patients with greater than 300 × 106/L CD34+ cells have more rapid progression of disease than patients with fewer CD34+ cells.289
Endothelial progenitor cells (CD34+CD133+ and VEGF receptor 2–positive cells) are significantly higher in the blood of primary myelofibrosis patients than of normal subjects.89
Mild lymphocytopenia resulting from decreased CD3+, CD4+, CD8+, and CD3–/CD56+ T cells is the rule.295
The neutrophils of some patients have impaired phagocytosis, oxygen consumption, nitroblue tetrazolium reduction, and hydrogen peroxide generation, and decreased myeloperoxidase296,297 and glutathione reductase activities.297 CD34+ cells have impaired in vitro differentiation to natural killer cells, which appears to be related to a dysregulation in control of IL-15.298
Bleeding time can be prolonged disproportionately to the platelet count.299,300 Platelet abnormalities include impaired aggregation in response to epinephrine, depletion of dense granule adenosine diphosphate content,301 decreased platelet lipoxygenase pathway activity,302 and others.303,304 The correlation of bleeding or thrombosis with platelet functional abnormalities is weak.303,304 The lupus anticoagulant has been present, rarely.242
In the fibrotic phase, marrow aspiration often is unsuccessful because of the fibrosis.8,9,10,11,12,13,14
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


