The primary immunodeficiency disease (PIDD) are a group of clinical syndromes originally described in patients with marked susceptibility to particular types of infections. This group of disorders has now expanded to include more than 150 clinically defined entities that span the full spectrum of immune dysfunction ranging from virtually absent immune responses to overwhelming uncontrolled autoimmunity.1 In this “genomic era” we have come to understand that the majority of immunodeficiency disorders are caused by defects in single genes. There are now approximately 200 known single gene defects associated with immune deficiency. It has also become evident that mutations in different genes can lead to a similar clinical phenotype. For example, defects in more than 20 different genes have now been associated with a clinical phenotype of severe combined immune deficiency (SCID).2 This has led to an explosion in our understanding of the role of specific molecular pathways in the human immune system but has created some confusion when discussing the clinical syndromes themselves. Consequently, it has become the practice to refer to disorders by their molecular defect, either in combination with, or in lieu of, their clinical name or eponym (i.e., “ADA-deficiency” or “ADA-SCID” rather than “SCID”). This practice is followed here.
MAJOR COMPARTMENTS OF THE IMMUNE SYSTEM
To provide a framework for this overview of immunodeficiency, it is worthwhile to divide the immune system into four major compartments: complement, phagocytes, B cells and antibodies, and T cells. The complement and phagocyte compartments are part of the “innate” arm of the immune system, whereas the B-cell and T-cell compartments are components of the “adaptive” arm of the immune system (see Table 63.1). Many immunodeficiency disorders are caused by a defect in only one compartment of the immune system, whereas others are “combined” immunodeficiencies with defects in multiple compartments. In general, defects in each compartment of the immune system are associated with susceptibilities to particular types of infections. In addition, each specific immunodeficiency typically has unique features that differentiate it from other immunodeficiencies, often making it possible to identify the disorders based on clinical and laboratory findings (see Table 63.1).
TABLE 63.1 COMMON SYMPTOMS ASSOCIATED WITH DEFECTS IN EACH OF THE FOUR MAJOR IMMUNE COMPARTMENTS
Complement
Phagocytes
B Cells/Antibodies
T Cells
Invasive infections with encapsulated bacteria (S. pneumoniae, H. influenzae, etc.)
Recurrent, invasive Neisserial infections
Hereditary angioedema
Autoimmunity (lupus, glomerulonephritis)
Familial hemolytic uremic syndrome (HUS)
Skin and soft tissue abscesses, boils, and lymphadenitis
Infections with Catalase+ organisms (S. aureus, Serratia, Aspergillus, etc.)
Poor wound healing
Chronic gingivitis and periodontal disease
Mucosal ulcerations, colitis
Delayed separation of the umbilical cord/omphalitis
Recurrent bacterial sinopulmonary infections
Unexplained bronchiectasis
Chronic or recurrent gastroenteritis (giardia, cryptosporidium, enterovirus, etc.)
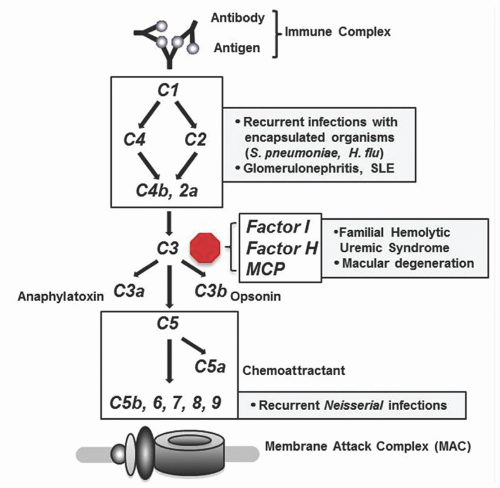
The complement system consists of a series of proteins that are present in the plasma and become activated upon encountering pathogens. Activation of early complement components initiates a cascade of protein cleavage and activation events that ultimately leads to formation of the membrane attack complex (MAC) consisting of complement proteins C5, 6, 7, 8, and 9. A number of regulatory proteins including Factor H, Factor I, and membrane cofactor protein (MCP) control complement activation at the level of C3, thereby preventing inappropriate complement fixation. The complement cascade is activated via three major mechanisms: (1) the classical pathway, which is initiated by antigen/antibody complexes; (2) the alternative pathway, which is initiated directly by bacterial cell wall components; and (3) the lectin pathway, which is initiated by carbohydrate moieties present on bacteria.
Complement deficiencies make up only a small portion (˜2%) of all primary immune deficiencies.3 Defective activation of the entire complement cascade can be caused by the absence or dysfunction of only 1 of more than 20 complement proteins. The proteins most often affected are C2, C3, and C4.4
Clinical Presentation
Patients with defects in the C1 esterase inhibitor have hereditary angioedema (HAE) in which allergic or mechanical stimuli can trigger massive, localized, severe attacks of edema that can be life-threatening if they involve the airway. Patients with deficiency of early complement components in the classical pathway (C1-C4) typically present with symptoms of autoimmunity (lupus or glomerulonephritis) or with recurrent invasive infections caused by encapsulated organisms (particularly Streptococcus pneumoniae). Patients with defects in the late complement components (C5-C9) that are involved in formation of the membrane attack complex typically present with recurrent or severe Neisserial infections.4 Patients with defects in complement regulatory proteins (Factor I, Factor H, and MCP) are at risk of developing familial hemolytic uremic syndrome and age-related macular degeneration (see Fig. 63.1).5, 6, 7
FIGURE 63.1. The classical complement pathway showing activation by immune complexes and ultimate formation of the membrane attack complex. Factor I, Factor H, and MCP (membrane cofactor protein) control complement activation primarily at the level of C3. Boxes indicate the clinical syndromes or infections that result from defects of the indicated complement proteins.
Specific Disorders
C1 Esterase Inhibitor Deficiency
Deficiency of C1 esterase inhibitor is the cause of HAE. Unlike many of the other early complement component deficiencies, absence of C1 esterase inhibitor does not lead to an increased risk for infection. Instead, this protein regulates the activity of kallikrein, blocking its ability to act on Factor XII and kininogen. As a consequence, minor irritants cause unabated production of bradykinin and other mediators of vascular permeability, leading to rapid swelling of the soft tissues (angioedema), severe abdominal pain, and at times, acute obstruction of the airway. Effective treatments are now available for HAE including purified C1 esterase inhibitor, a kallikrein inhibitor, and a bradykinin B2 receptor antagonist.8, 9, 10 Because of the expense of these agents, they are often administered at the beginning of an attack to abort symptoms rather than being administered prophylactically to prevent the onset of an attack.
C2 Deficiency
C2 deficiency is the most common complement component deficiency associated with susceptibility to infections, occurring in approximately 1 of 20,000 people. As indicated above, patients with early complement cascade defects (C1-C4) are susceptible to invasive infections with encapsulated organisms, S. pneumoniae being a particularly fulminant pathogen in these patients. The infectious susceptibility is compounded by functional antibody deficiency in some patients. In addition, patients with C1, C2, or C4 deficiency are at high risk of developing autoimmunity (lupus or glomerulonephritis). In the case of homozygous C2 deficiency, approximately 50% of patients develop lupus or glomerulonephritis.11
Diagnosis
Screening for complement deficiency should be performed in patients with recurrent episodes of bacteremia, meningitis, or disseminated gonorrhea. Because more than 30% of patients with recurrent Neisseria infections have complement deficiency, it is imperative that prompt testing of the complement system be done in these individuals. The screening test of choice for complement deficiencies is the CH50 test, which measures functional complement activity in the plasma. The CH50 will only identify defects in the classical pathway, which is typically sufficient inasmuch as alternative pathway defects are exceedingly rare. If an alternative pathway defect is suspected, however, an analogous test (the AH50) can be performed. In order for the CH50 to give accurate results, the blood specimen needs to be handled carefully because complement is very heat labile. In general, it is recommended that any abnormal CH50 test should be repeated to confirm a complement deficiency. The CH50 is typically very low or absent in patients with a complement component deficiency. A CH50 result that is only moderately low is often seen in situations where the specimen was handled incorrectly or in patients with autoimmune disease such as lupus or mixed connective tissue disease. Once an abnormal CH50 test is confirmed, specialized testing to identify the specific complement component that is absent can be performed.
Treatment
Patients with complement deficiency are susceptible to fulminant sepsis and other deep-seated infections caused by encapsulated organisms. For this reason, patients should be given a letter or laminated card that they can keep with them at all times with contact information for their primary care physician and clinical immunologist, and a message indicating that they have a complement deficiency and there should be no delay in giving parenteral antibiotics should they be ill. For patients who live at a distance from skilled medical care, consideration should be given to providing a dose of parenteral antibiotic such ceftriaxone that can be administered by the patient or a family member when they become ill, prior to a lengthy trip to the hospital. The efficacy of chronic prophylactic antibiotics to prevent infection in patients with complement deficiency is not well studied and remains a significant question in this group of disorders. In addition to preparing for and treating infections, patients should also be regularly screened for autoimmunity by history, physical exam (i.e., blood pressure monitoring), and laboratory testing including BUN, creatinine, and urinalysis to monitor for signs of glomerulonephritis inasmuch as this is a common autoimmune manifestation.
PHAGOCYTES
One of the major roles of phagocytic cells (neutrophils and macrophages) is to continuously survey the body for signs of infection. Upon sensing an infection, they migrate from the circulation into the tissues toward the site of the infection where they begin to ingest both opsonized and nonopsonized pathogens and debris. The ingested material is processed and fragments of digested proteins are loaded into class II MHC molecules that are presented at the cell surface where they can be recognized by cells of the adaptive immune system. Phagocytes that ingest pathogens and debris can either remain at the site of infection or migrate back to local draining lymph nodes to present antigen. Phagocytic disorders can occur in one of three ways: (1) a lack of phagocytes (congenital neutropenia); (2) defective phagocyte migration (leukocyte adhesion deficiency [LAD] and WHIM syndrome); and (3) inability of phagocytes to process or degrade organisms that have been ingested (chronic granulomatous disease [CGD]).
Clinical Presentation
Because of the role that phagocytes play in controlling bacterial and fungal pathogens, patients with phagocytic defects often present with infections and abscesses of skin, deep tissues, and organs caused by bacteria and fungi. Symptoms can include boils and/or cellulitis with or without pus, lymphadenitis, pneumonia, delayed shedding of the umbilical cord, hepatic abscesses, gastrointestinal disorders, gingivitis, and unexplained fever, malaise, and fatigue. The onset of symptoms of phagocytic cell disorders is typically in infancy or early childhood.
Specific Disorders
Severe Congenital Neutropenia
The congenital neutropenias are described in more detail elsewhere (Chapter 57) so are covered only briefly here. Mutations in five different genes have now been associated with severe congenital neutropenia (SCN): ELANE, which is inherited in an autosomal dominant manner, causes increased myeloid cell apoptosis and can present either with SCN or with a cyclic neutropenia phenotype; GFI1, which is also inherited in an autosomal dominant manner, causes defective myeloid cell differentiation; HAX1, which is inherited in an autosomal recessive manner, is associated with increased myeloid cell apoptosis, and is the cause of the classic “Kostmann” syndrome; G6PC3, which is inherited in an autosomal recessive manner, causes excessive myeloid cell apoptosis, and is associated with a variety of other congenital defects including cardiac, urogenital, endocrine, auditory, and facial anomalies; and WAS, in which specific activating mutations in the CDC42 binding domain are inherited in an X-linked recessive manner, leading to abnormal and dysregulated actin polymerization that causes defective neutrophil chemotaxis and increased apoptosis.12
In all cases, clinical management involves a heightened suspicion for infections and aggressive treatment if these arise. Treatment of acute infections may require antibiotics combined with G-CSF to increase neutrophil counts. Despite there being little evidence specifically in SCN supporting the use of prophylactic antibiotics, extrapolation from data in leukemic patients with neutropenia suggests a benefit so these are used in most patients. Prophylactic, long-term therapy with G-CSF is typically utilized only in those patients who have recurrent, severe bacterial infections in spite of antibiotic prophylaxis or in patients with fungal infections. Bone marrow transplantation is effective in SCN although there is little to no reported experience in those genetic disorders that are more rare such as G6PC3 deficiency.
Leukocyte Adhesion Deficiency
LAD is caused by the absence of functional adhesion receptors that are required for the migration of phagocytes from the circulation into the tissues. The characteristic clinical features of LAD include recurrent skin and soft tissue infections that often lead to development of deep ulcers despite there being highly elevated peripheral blood leukocyte counts. Interestingly, the inability of leukocytes to migrate to these sites of infection leads to an absence of pus in the lesions, which can be a useful diagnostic clue. Wound healing is also compromised and patients typically have marked gingivostomatitis. Three forms of LAD have been described: LAD-I, the most common form of LAD, is caused by mutations in the ITGB2 gene encoding the β2-integrin CD18. Mutations cause an absence of the CD11/CD18 integrin complex on the surface of leukocytes, which can be readily discerned by flow cytometry. LAD-II is caused by mutations in the SLC35C1 gene encoding the GDP-fucose transporter. These mutations cause defective expression of Sialyl Lewis X (sLeX), a fucose-containing ligand on neutrophils. sLeX is the ligand for E- and P-selectins, which are expressed on the surface of cytokine-activated endothelial cells and allow neutrophil rolling. As a result of the fucose defect, all patients with LAD-II also have the rare Bombay blood group, which is a useful diagnostic test for suspected LAD-II. LAD-III is caused by mutations in the FERMT3 gene that encodes Kindlin-3, a coactivator that is required for activation and function of β1-, β2-, and β3-integrins. Absence of functional Kindlin-3 leads to dysfunction of CD18 and causes an LAD phenotype (see LAD-I above). In addition, patients with LAD-III also have a Glanzmann-type bleeding disorder resulting from dysfunctional integrin-mediated aggregation of platelets.13
Patients with LAD-I and LAD-III typically present in childhood and often have a severe course with early mortality, whereas patients with LAD-II often have milder cases and may live into adulthood. Treatment of LAD can be more complicated than some of the other phagocytic disorders because in addition to treating infections aggressively with antibiotics, active soft tissue infections may require recurrent donor white cell infusions of functional neutrophils in order to clear the infection. The primary defects of LAD are intrinsic to hematopoietic cells, thus bone marrow transplantation can be curative.14, 15, 16
WHIM Syndrome
WHIM syndrome (warts, hypogammaglobulinemia, recurrent bacterial infections, and myelokathexis [retention of neutrophils in the bone marrow]) is caused by autosomal dominant mutations in CXCR4, the receptor for the chemokine CXCL12 (SDF-1). Patients with WHIM typically present in childhood with recurrent bacterial otitis media, sinusitis, bronchitis, pneumonia, and cellulitis. The bacterial susceptibility is a result of the combination of hypogammaglobulinemia and neutropenia. In addition to bacterial infections, patients with WHIM have a particular susceptibility to papillomavirus infections, which can be severe and lead to early malignancy. The mechanisms that underlie the viral susceptibility are not entirely understood but are thought possibly to be intrinsic to the epithelial cells. In the hematopoietic system, CXCL12 causes homing of cells to the bone marrow and controls release of these cells from the marrow. Neutrophils and lymphocytes from patients with WHIM have an increased chemotactic response to CXCL12, suggesting that the neutropenia and lymphopenia observed in WHIM are the result of inappropriate cell retention in the marrow.17 Treatment with G-CSF or GM-CSF can normalize the neutrophil counts although these often cause significant bone pain at the doses required.18 Recent studies using the CXCR4 antagonist plerixafor in adults with WHIM syndrome have shown promise for improving neutrophil counts by mobilizing neutrophils from the bone marrow.19, 20 Antibiotics and immunoglobulin replacement can significantly reduce the risk of bacterial infections. There is very little reported experience regarding bone marrow transplantation for WHIM although anecdotal evidence suggests that it may correct the neutropenia and hypogammaglobulinemia but may not alter the papillomavirus susceptibility.21
Chronic Granulomatous Disease
CGD is the most frequently diagnosed phagocytic cell immune defect. The most common form is X-linked, caused by mutations in the CYBB gene and accounting for approximately two thirds of all CGD patients. The remaining forms, caused by mutations in the CYBA, NCF1, NCF2, or NCF4 genes are all autosomal recessive. All mutations affect the formation or function of the NADPH oxidase complex, on neutrophil phagolysosomes. The NADPH oxidase is required to generate a burst of reactive oxygen species in response to phagocytosis of pathogens. Reactive oxygen species activate proteases in the phagolysosomes that destroy ingested bacteria. In CGD, the oxidative burst cannot be generated, leading to defective processing of ingested organisms and an inability to eliminate bacterial and fungal pathogens appropriately. Catalase-positive organisms including Staphylococcus species, Aspergillus species, Burkholderia cepacia, Serratia marcescens, and others are the most common pathogens. The most common types of infection at presentation are pneumonia, lymphadenitis, cellulitis, and hepatic abscesses (particularly with Aspergillus sp.). The most common cause of premature death is Aspergillus infection.22 In addition to the infectious susceptibilities of CGD, a substantial percentage of patients struggle with inflammatory complications that are common to this disorder including an inflammatory colitis that occurs in approximately 40%, hepatic dysfunction, gingivitis, and others.
Treatment of CGD revolves around aggressive management of acute infections followed by prophylaxis against future infections using a combination of daily antibiotic (typically trimethoprim-sulfamethoxazole), daily antifungal (typically itraconazole), and thrice-weekly Interferon-γ injections. This combination has dramatically improved outcomes in CGD; however, despite appropriate use of this regimen, some patients ultimately have increasing symptoms that lead to a decline in survival beginning in the late teens or early twenties. The prospects for long-term survival appear to be correlated with the amount of residual oxidative burst activity that can be generated by a particular patient’s phagocytes.23 This has led to a renewed interest in bone marrow transplantation for CGD, which has been quite successful in the modern era, likely due to improved antimicrobials and transplant-conditioning regimens with reduced toxicity. Many now recommend that for patients who have mutations that severely affect oxidative burst activity, bone marrow transplant should be considered preemptively, early in life before patients develop comorbidities.24
Diagnosis
Assessment of patients for a possible phagocytic disorder requires that both the number and the function of phagocytes be evaluated. Numbers are easily evaluated using a complete blood count with differential. If there is a concern for cyclic neutropenia, neutrophil counts may need to be evaluated 3 times weekly for 3 to 4 weeks to identify the nadir.25 Functional testing includes evaluation of CD11/CD18 integrin expression on myeloid cells by flow cytometry if the patient has symptoms suggestive of leukocyte adhesion deficiency. In cases of suspected CGD, evaluation of neutrophil oxidative burst function is essential. Traditionally this was done using nitroblue tetrazoleum (NBT) but is now performed using dihydrorhodamine (DHR), a reagent that permeates neutrophils and fluoresces when reduced by a normal neutrophil oxidative burst. Fluorescence is measured by flow cytometry. The DHR test is sensitive enough to differentiate most cases of X-linked CGD from autosomal recessive CGD, making it a particularly useful clinical assay.26, 27
Treatment
As noted above, management of phagocytic disorders revolves around having a heightened suspicion for infections, aggressively treating acute infections using antibiotics and G-CSF as necessary, and developing a prophylaxis regimen that is both effective and reasonable from a patient standpoint. At times, patients continue to have recurrent or severe infections despite these efforts and require more definitive therapy. As indicated above, hematopoietic stem cell transplantation (HSCT) has been shown to be effective in many, but not all, phagocytic disorders. Gene therapy has been attempted for both X-linked CGD28 and for LAD-I29 but neither has been particularly successful thus far and at this point is considered to be experimental. Ongoing research to address the challenges of gene therapy that are unique to these two disorders is underway.
B CELLS/ANTIBODIES
The predominant role of B cells in the immune system is to make antibodies (immunoglobulins) in response to antigen challenge (pathogens, vaccines, etc.). The absence of functional antibodies causes susceptibility to bacterial and viral infections. Antibody deficiency can occur in one of three different ways: (1) hypogammaglobulinemia or low levels of one or more immunoglobulin classes (IgG, IgA, IgM, or IgE) occurring as a result of decreased antibody production, often associated with a specific single-gene defect; (2) hypogammaglobulinemia as a result of excessive antibody loss, typically through the kidneys as proteinuria or through the gut as protein-losing enteropathy; or (3) functional antibody deficiency, in which immunoglobulin levels are normal but the Ig lacks the quality required to bind and opsonize pathogens.
Clinical Presentation
Patients who lack sufficient levels of functional antibody present clinically with recurrent bacterial sinopulmonary infections (sinusitis, otitis media, bronchitis, and pneumonia). In addition, patients may develop bowel infections caused by microorganisms such as Giardia or Cryptosporidium that are often only modestly pathogenic to normal individuals. In addition to these symptoms, patients with certain antibody-deficiency disorders have characteristic clinical features that can provide clues to the specific diagnosis. These are covered below.
Specific Disorders
X-Linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA) is caused by mutations in the Bruton’s tyrosine kinase (BTK) gene. BTK is a member of the Tec family of cytoplasmic tyrosine kinases and is required for the maturation of B-cell precursors in the bone marrow.30 Mutations in BTK therefore cause an arrest of B-cell development at the pre-B-cell stage leading to virtual absence of circulating B cells in the peripheral blood. Mutations that only partially interfere with the enzymatic function of BTK have also been described and are associated with milder forms of the disease that only have defects in the generation of specific antibody responses.
XLA is typically suspected in male patients with recurrent bacterial sinopulmonary infections and <2% circulating CD19+ B cells. Other infections that occur relatively frequently in patients with XLA prior to the initiation of IgG replacement therapy include skin infections (furunculosis, pyoderma, and cellulitis) and sepsis. The diagnosis can be confirmed either by identifying a mutation in the BTK gene or by demonstrating absence of the BTK protein in monocytes or platelets. A positive family history suggestive of an X-linked recessive mode of inheritance increases the suspicion for XLA. It is uncommon for patients with XLA to develop symptoms in the first months of life because newborns are protected from most infections by transplacentally acquired maternal IgG. There are few distinguishing physical features of XLA that can provide clues to the diagnosis but absence of visible tonsils or adenoids (by X-ray or CT scan) is a useful clue.
In addition to the common sinopulmonary pathogens, patients with XLA are also susceptible to infections by particular opportunistic organisms that are more rare and fastidious, which can cause unusual clinical syndromes. For example, Helicobactercinaedi can cause a syndrome of dermatomyositis and cellulitis that presents with cutaneous ulcerations, particularly on the lower legs.31, 32 The organism can sometimes be recovered from the blood but is fastidious and difficult to culture using usual methods. The ability to culture the organism and evaluate its antibiotic sensitivity is crucial in many cases because it is frequently resistant to various antibiotics. A combination of antibiotics is often needed to clear the infection effectively.33 Similarly, Mycoplasma species, including M. hominis, can cause lung, abdominal, or bone infections that are remarkably hard to eradicate and Ureaplasma urealyticum infections are a rare cause of arthritis, urethritis, and pneumonia.34
Prior to the widespread use of IgG supplementation in these patients, opportunistic viral infections, particularly with viruses that require an extracellular phase, were especially problematic. For example, echovirus encephalitis was estimated to be the cause of death in ˜10% of boys with XLA in the 1970s but that number has fallen dramatically with aggressive use of IgG supplementation. There continue to be rare cases of echovirus encephalitis in patients with XLA, even in those on adequate IgG replacement therapy, but these are thought to be caused by viral strains for which there may not be high antibody titers in the particular IgG preparation being used.35, 36 Similarly, a mink astrovirus strain was recently identified by deep sequencing from the brain of an XLA patient who developed a neurodegenerative disorder as a teen.37 Interestingly, he had lived next to a mink farm as a child but was started on immunosuppression early in his teens for inflammatory bowel disease, which may have allowed the virus to escape control. Lastly, infections with vaccine-strain poliovirus were pathogenic in undiagnosed patients with XLA who were immunized with the live-viral vaccine after transplacentally acquired maternal antibody had waned. The shift from live attenuated to killed poliovirus for immunization has essentially eliminated new cases.
Hyperimmunoglobulin M Syndromes
Under normal circumstances, binding of antigen to cell surface IgM on naive B cells induces activation of the B-cell. The antigen that is bound to surface IgM on the B-cell is ingested, proteolytically digested, and antigenic peptide fragments are displayed on the B-cell surface in MHC Class II molecules. Antigen-specific T cells then engage the B-cell via MHC II/TCR interactions. Once engaged, the activated helper T-cell (Th) provides additional costimulatory signals to the B-cell that are critical to promote immunoglobulin class-switching from IgM to IgG, IgA, and IgE. The most important of these costimulatory signals comes via the interaction of CD40 ligand on activated T cells with CD40 on B cells. Additional costimulatory signals come from ICOS on activated T cells and ICOS ligand (B7-H2) on B cells. Activation of the B-cell through CD40 and cytokines that are secreted by the helper T-cell cause it to undergo class-switch recombination (CSR) during which the µ-heavy chain gene segment within the immunoglobulin gene, is replaced by either a γ, α, or ε gene segment. This is accomplished by nicking and double-strand breakage of the DNA in the immunoglobulin heavy-chain locus, which requires a series of enzymatic steps that involve activation-induced cytidine deaminase (AID), uracil DNA glycosylase (UNG), and others. Genetic defects that affect CD40 ligand (CD40L), CD40, AID, or UNG can therefore prevent class-switch recombination thus thwarting the B-cell’s ability to make significant amounts of any antibody isotype in addition to IgM.
The overwhelming majority of patients with hyper-IgM syndrome have the X-linked form caused by X-linked recessive mutations in the CD40 ligand (CD40L/CD154), which is encoded by the CD40L gene on the X-chromosome.38 CD40L and its receptor CD40 are members of the tumor necrosis factor (TNF) superfamily of ligands and receptors. In lymphocytes, CD40L is expressed only on activated T cells but is also expressed on platelets where its role is unknown. Affected boys may present with recurrent bacterial sinopulmonary infections caused by low IgG, IgA, and IgE, whereas IgM is normal or elevated. In addition to the usual bacterial pathogens, patients with CD40L mutations also demonstrate unique susceptibilities to fungal infections, particularly Pneumocystis jiroveci (PJ) that causes pneumonia, and to a protozoan, Cryptosporidium parvum (CP) that causes bowel infections. B lymphocytes are present, and T-cell numbers are generally normal. In almost all cases, the diagnosis can be made by using flow cytometry to evaluate the expression and function of the CD40L protein on activated T cells. Expression is evaluated using antibodies specific to the CD40L protein and the function is evaluated by measuring the binding of a CD40-Ig heavy chain fusion protein to the expressed CD40L. Gene sequencing can then be performed in order to identify a specific mutation.
The susceptibility to Pneumocystis and possibly other fungal pathogens has been somewhat of a puzzle because patients do not have other signs of a significant cellular immune defect such as severe or recurrent viral infections. Interestingly, the susceptibility to Pneumocysitis jiroveci pneumonia (PJP) appears to go away in most patients by the age of 5. PJP is almost always diagnosed by staining bronchoalveolar lavage fluid for the presence of the organism. PJP can be readily prevented by prophylactic trimethoprim-sulfamethoxazole administration and active disease is amenable to treatment using higher doses of the same drug. Recent data have suggested that the fungal susceptibility in CD40L deficiency may be a result of defective CD40L signaling into dendritic cells and monocytes that express CD40.39
C. parvum bowel infections are more difficult to diagnose and manage in patients with CD40L deficiency. CP may cause abdominal pain, bloating, diarrhea, malabsorption, and weight loss. It may require multiple stool samples to identify the oocysts and occasionally, the diagnosis can only be made on endoscopically obtained biopsy specimens. Treatment with paromomycin or nitazoxanide can clear the infection although prolonged courses are typically needed in hyper IgM patients and treatment failures are not uncommon. CP infections can result in chronic inflammation of the gut and biliary tree, which seems a likely contributor to the high incidence of bile duct cancers seen in these patients.40, 41
Treatment involves the use of IgG replacement therapy combined with prophylactic antibiotics for prevention of PJP at least until the age of 5. The role of bone marrow transplantation for CD40L deficiency is still being evaluated. Even though a number of patients have undergone successful bone marrow transplantation, the role of BMT remains somewhat controversial in this disease although in patients with ongoing CP infection, the prognosis for the patients who develop bile duct disease is so poor that the risks of transplantation are well justified.42, 43
CD40 deficiency is inherited as an autosomal recessive defect that has been described primarily in two cohorts from Italy and the Middle East. It results in a syndrome that is almost identical to CD40L deficiency in which both sexes are affected. Homozygosity for a null mutation occurs, but heterozygosity is more common and can result in a partial phenotype because CD40 functions as a trimer (defects of even one chain of the trimer interfere with its function).44
Autosomal recessive mutations in AID and UNG also cause a hyper-IgM phenotype but it tends to be milder than either CD40L or CD40 deficiency, likely because the defect is limited to B cells, whereas CD40L/CD40 signaling plays a role in other cell types including dendritic cells and monocytes.45, 46 Patients with AID or UNG typically live into adulthood and do not demonstrate the same susceptibility to PJP and CP bowel infections. Patients with mutations in AID do, however, have a significant propensity to develop autoimmunity affecting various organ systems. Patients are typically treated with IgG replacement therapy and antibiotics for acute infections. There are no reports of bone marrow transplantation for AID or UNG deficiency.
Common Variable Immunodeficiency Syndromes
Common variable immunodeficiency (CVID) is a heterogeneous disorder that is likely caused by a variety of molecular mechanisms that ultimately lead to a similar clinical phenotype. The European Society of Immunodeficiency (ESID) has proposed diagnostic criteria in an effort to standardize the diagnosis of CVID. These include: (1) plasma IgG levels that are less than 2 standard deviations below the mean for age combined with a “marked decrease” in either IgM or IgA; (2) age of onset of immunodeficiency >2 years of age; (3) absent isohemagglutinins or poor responses to vaccines; and (4) defined causes of hypogammaglobulinemia have been excluded.
The peak age of onset of CVID is in the second or third decade of life and 50% to 60% of patients have a clinical phenotype consisting almost exclusively of increased bacterial sinopulmonary infections. With IgG supplementation, this group of patients has a relatively benign course with long-term survival that is not unlike the normal population. The other half of patients have a complicated disease course with autoimmunity or lymphoproliferative disease that can involve the hematopoietic system, lungs, lymph nodes, liver, and bowel. The long-term outcome of this population is significantly worse, approaching only 40% survival over 40 years.47
Among the disorders seen in this population, a granulomatous, lymphoproliferative, interstitial lung disease (GLILD) affects approximately 30% to 40% of patients.48 This often presents with decreasing lung function that is manifested by cough, decreased exercise tolerance, and sometimes hypoxemia. Typical findings on chest CT scan include diffuse nodules within the lung, opacities that have a “ground glass” appearance, bronchial wall thickening, and sometimes bronchiectasis. Lung biopsy generally demonstrates a lymphocytic interstitial pneumonitis with noncaseating granulomas and a follicular bronchiolitis with lymphoid aggregates of both B and T cells. This pattern is sometimes mistaken for sarcoidosis although there are differences. Over time, this inflammatory process in the lungs will cause destruction of alveoli and will contribute to development of bronchiectasis. There continues to be some debate among providers about whether this process should be treated if the patient is not demonstrating pulmonary compromise. If left unchecked, however, there is evidence that irreversible damage and fibrosis develops in many patients. High-dose steroids are often used as first-line therapy to treat this process and in many cases they are effective but do not typically lead to a lasting remission on their own. A recent study using a combination of anti-CD20 monoclonal antibody (rituximab) therapy and azathioprine in a small cohort of CVID patients with GLILD demonstrated dramatic responses with a prolonged remission of disease in many patients.49
In addition to pulmonary symptoms, gastrointestinal complaints are common in CVID, affecting 20% to 30% of patients.47 Patients who develop disease demonstrate a hypertrophic lymphoproliferation of Peyer’s patches that causes a nodular lymphoid hyperplasia in the bowel. This is associated with abdominal discomfort, diarrhea, malabsorption, and weight loss and can cause significant morbidity. A variety of approaches have been taken to treat this process but none have offered particularly dramatic results although nonabsorbable steroid preparations have shown some benefit with minimal side effects. A more troubling complication observed in 5% to 10% of patients is a hepatitis that can cause severe hepatic dysfunction with development of hepatosplenomegaly and ascites.47 Infectious causes are almost never identified and liver biopsy demonstrates a nodular lymphoid hyperplasia in the liver parenchyma, not unlike that observed in the bowel. Liver disease is among the complications associated with a poor outcome.
Some 20% of subjects have additional clinical findings that are suggestive of autoimmunity/immune dysregulation including immune thrombocytopenia and hemolytic anemia, neuropathy, endocrinopathies, and skin disease. Skin involvement ranges from alopecia and vitiligo to psoriasis and granuloma annulare.50, 51
Only gold members can continue reading. Log In or Register to continue
 The Diagnostic and Therapeutic Approach to Hematologic Problems
The Diagnostic and Therapeutic Approach to Hematologic Problems



