Primary and metastatic neoplasms of the brain in adults
Lisa M. DeAngelis, MD, FAAN
Overview
Primary brain tumors are uncommon but aggressive neoplasms even when histology is low grade. Secondary, or metastatic, tumors are at least 3 times more common than all primary tumors combined. Diagnosis is easily established by magnetic resonance imaging (MRI), and surgical resection provides histologic confirmation and decompression; surgery alone may suffice for low-grade tumors and most extra-axial tumors such as meningiomas. Treatment of parenchymal brain tumors, primary or metastatic, often requires a combination of radiotherapy and chemotherapy, which may improve patient function and control disease but rarely achieves cure.
Introduction
Tumors of the central nervous system (CNS) are a heterogeneous group of both benign and malignant intracranial neoplasms. CNS tumors are classified into two groups: those that grow within the brain (intracerebral) and those that grow outside the brain (extracerebral). Historically, CNS tumors have been intractable to standard therapies of surgical resection, radiotherapy (RT), and chemotherapy. However, over the past several decades, advances in diagnostic imaging, surgical techniques, radiation oncology, and chemotherapy have improved survival and quality of life. Most important, we are acquiring a better understanding of the molecular events associated with the malignant phenotype of a brain tumor, which has led to several novel chemotherapeutic approaches to their treatment.
Epidemiology
Primary intracranial tumors are uncommon. The overall annual incidence of brain tumors, both benign and malignant, in the United States is estimated at 21.03 cases per 100,000 person-years leading to an estimated 63,000 new cases diagnosed each year (Central Brain Tumor Registry of the United States Statistical Report [CBTRUS] 2006–2010).1 More than 26% are gliomas, and almost three-quarters of these are high grade with a higher incidence in men (7.76 per 100,000 person-years) than in women (5.60 per 100,000 person-years).1
The incidence and histologic type of intracranial tumor differs by race, gender, and age.1 The overall incidence of brain tumors (especially gliomas) is greater in Whites than Blacks. However, meningiomas are more frequent in Blacks than Whites. Pituitary adenomas are also more common in Blacks than Whites. Gender differences are apparent. The male to female ratio is 1.3 for oligodendrogliomas, 1.38 for astrocytomas, and 0.77 for malignant meningiomas, and for benign meningiomas, the male to female ratio is 0.44. Lymphomas and germ cell tumors are more common in males, 1.31 and 2.33, respectively.
CNS tumors can occur at any age, but the incidence and histologic type vary by age. There is a small peak before age 10 and a steady rise from age 15. The average age of onset for all primary brain tumors is 54 years. The average age of onset of glioblastoma (GBM) and meningioma is 62 years, whereas for oligodendroglioma the mean age of onset is 16 years. The CBTRUS data show that the highest incidence for all brain tumors occurs in the 75- to 84-year-old age group, with GBMs occurring most frequently in patients older than 65 years. Survival is directly related to patient age and tumor histology. For instance, the 5-year relative survival rate for patients with pilocytic astrocytomas (a common childhood tumor) is 94.4%, compared to 4.7% in patients with GBMs. Other important prognostic factors include extent of disease, extent of resection, and tumor location.
Risk factors
There have been a large number of studies examining the relationship between the environment and the occurrence of brain tumors, but only two unequivocal risk factors have been identified: ionizing radiation and immune suppression (Table 1). The role of low- and high-dose therapeutic ionizing radiation as a significant risk factor for brain tumors has been confirmed in many studies. Irradiation for intracranial tumors, e.g., medulloblastoma or extracranial head and neck cancers, including prophylactic irradiation for leukemia, increase the incidence of both gliomas and sarcomas sevenfold in those who survive more than 3 years. The cumulative relative risk of secondary brain tumors in patients treated with cranial irradiation ranges from 5.65 to 10.9; approximately two-thirds of the tumors are gliomas and one-third are meningiomas.2 High-grade gliomas have a median latency of 9.1 years from cranial RT compared to 19 years for meningiomas. Low-dose radiation such as that used to treat tinea capitis, a fungal infection of the scalp, and skin hemangiomas in children is associated with an increased risk of brain tumors. A relative risk of 18, 10, and 3 have been observed for nerve sheath tumor, malignant meningioma, and glioma, respectively.
Table 1 Risk factors for primary brain tumors of neuroepithelial, meningeal, or lymphocytic origin
| Definite risk factors |
| Ionizing radiation |
| Hereditary syndromes |
| Family history of brain tumors |
| Immunosuppression |
Congenital or acquired immune suppression, such as human immunodeficiency virus (HIV) infection, or the use of immunosuppressive drugs after organ transplantation, increases the incidence of primary central nervous system lymphoma (PCNSL).3 HIV infection may also increase the frequency of glioma and intracranial leiomyosarcomas. PCNSL in immunosuppressed patients is driven by a pre-existing latent Epstein–Barr viral infection of B-lymphocytes. When a lymphoma occurs in an immunosuppressed patient, it is twice as likely to occur in the brain as elsewhere in the body.
Other studies of environmental risk factors are less convincing than those of ionizing radiation and immunosuppression, including dietary exposure to N-nitrosourea compounds, occupational exposure to pesticide and fertilizer manufacturing, formaldehyde, synthetic rubber production, vinyl chloride synthesis, and petrochemical industries. Additional research evaluating the association of brain tumors with head trauma, cigarette smoking, seizure history, maternal alcohol use, and infection has been inconclusive.
Exposure to electromagnetic fields (EMF), especially through the use of cellular phones, has been of interest. Most studies have demonstrated no association between cellular phone use and brain tumors, but there may be a dose relationship to brain tumor risk.4, 5 Recent data suggest a history of allergies or atopic disease may be protective against glioma development. In addition, large genome-wide association studies point to eight germline DNA single nucleotide polymorphisms in seven genes that predispose to glioma formation or specific glioma grade or subtypes; the mechanism of this predisposition is unknown.6
Molecular genetics
There is evidence that somatic mutations in different molecular pathways may lead to the development of an identically appearing GBM. There appear to be at least four different molecular subclasses of GMB that include (1) classical characterized by EGFR amplification/mutation and p53 mutation, (2) proneural characterized by alterations in PDGFRA and isocitrate dehydrogenase (IDH) mutations, (3) mesenchymal characterized by NF1 dysfunction, and (4) neural with EGFR overactivity and displaying neural markers.7 At the present time, this molecular classification scheme does not drive treatment selection or prognosis. Lower grade gliomas with either astrocytic or oligodendroglial features are characterized by IDH mutations, primarily the IDHR132H mutation in 70%.8 This mutation leads to the formation of the oncometabolite, 2 hydroxyglutarate (2HG), which has become both a diagnostic and therapeutic target using magnetic resonance (MR) spectroscopy and novel IDH1 inhibitors. IDH1 mutations are thought to arise early in gliomagenesis, but they are noticeably lacking in primary GBMs, or those that present in older age as de novo GBMs.
In addition to oncogene amplification and tumor suppressor gene inactivation, malignant tumors acquire the ability to invade adjacent neural structures and form a new blood supply via angiogenesis. Glial tumors are extremely invasive neoplasms and typically extend well beyond their macroscopic or imaging borders. Angiogenesis leads to vascular hyperplasia, which is the pathologic hallmark of a GBM. Vascular endothelial growth factor (VEGF) is one of the key stimulants to angiogenesis in gliomas. VEGF is not expressed in normal brain endothelium but is upregulated in gliomas by local hypoxia. VEGF also increases vascular permeability and plays a role in the peritumoral edema seen in malignant gliomas.
Familial tumor syndromes of the CNS
Familial brain tumor syndromes are a heterogeneous group of disorders characterized by an association of brain tumors with systemic features, primarily dermatologic (Table 2).
Table 2 CNS tumor syndromes
| Disorders | CNS tumors | Tumors of other organs and tissues | Skin lesions | Genes | Chromosomes |
| Neurofibromatosis-1 | Glioma, neurofibroma | Iris hamartoma, osseous lesions, pheochromocytoma, leukemia | Café au-lait spots, cutaneous axillary freckling, neurofibromas | NF1 | 17q11.2 |
| Neurofibromatosis-2 | Vestibular Schwannoma, Meningioma | Posterior lens opacities, retinal hamartoma | None | NF2 | 22q12.2 |
| von Hippel–Lindau disease | Hemangioblastoma | Retinal hemangioblastoma, renal cell carcinoma, pheochromocytoma, visceral cysts, endolymphatic sac tumor | None | VHL | 3p25-p26 |
| Tuberous sclerosis | Subependymal giant cell astrocytoma (SEGA) | Cardiac rhabdomyoma, adenomatous polyps of the duodenum and small intestine, cysts of the lung and kidney, lymphangioleiomyomatosis, renal angiomyolipoma | Cutaneous Angiofibroma (“adenoma sebaceum”), peau de chagrin, subungual fibromas | TSC1, TSC2 | 9q34.14, 16p13.3 |
| Li–Fraumeni syndrome | Gliomas (10%) | Breast carcinoma; bone and soft tissue sarcoma; adrenocortical, lung, and GI carcinoma; leukemia | None | P53 | 17p13.1 |
| Cowden disease | Cerebellar mass (Lhermitte Duclos disease) | Hamartomatous polyps of the eye, colon, and thyroid; breast carcinoma, thyroid cancer | Multiple trichilemmomas, fibromas | PTEN | 10q22.3 |
| Brain tumor-polyposis syndrome type 1 | Malignant glioma | Colorectal adenomas, colon carcinoma, No polyps | MLH1 MSH2 MSH6 PMS2 | 3p21.3 2p21-22 2p16 7p22.1 | |
| Brain tumor-polyposis syndrome type 2 | Medulloblastoma | Colon cancer, colonic polyps | APC | 5q21 | |
| Nevoid basal cell carcinoma syndrome (Gorlin syndrome) | Medulloblastoma (anaplastic) | Jaw cysts, ovarian fibromas, skeletal abnormalities | Multiple basal cell carcinomas, palmar and plantar pits | PTCH | 9q22.3-31 |
| Retinoblastoma | Pineal tumor | Retinal tumor, osteosarcomas and other tumors | None | RBI | 13q14 |
| Bloom syndrome | Medulloblastoma meningioma | Characteristic face and voice, gonadal failure, diabetes, immunodeficiency | Sun sensitivity, patches of hyper-and hypopigmentation | BLM | 15q26.1 |
| Fanconi anemia | Astrocytoma, medulloblastoma | Anemia, skeletal malformations, enlarged cerebral ventricles, gastrointestinal malformations, AML | Café au-lait spots, hyper- and hypopigmentation | FANCA | 16q24.3 |
| Familial melanoma | Astrocytoma | Melanoma, pancreas, breast | Nevi | CDKN2A p16(1NK4) | 9p21.3 |
| Rhabdoid predisposition syndrome | PNET, choroid plexus carcinoma | Renal tumors, extrarenal malignant rhabdoid tumors | None | HSNFA/INH1 | 22q11 |
| Multiple endocrine neoplasia (MEN-1 Carney complex) | Pituitary adenomas | Hyperparathyroidism, gastrinoma, insulinoma, thyroid/bronchial carcinoid | Facial angiofibroma, lipoma, collagenoma | MEN1 | 11q13 |
| Ataxia-telangiectasia | Astrocytoma, medulloblastoma, cerebellar ataxia | Lymphomas, hypogonadism, radiationsensitivity, insulin resistance, premature aging, small stature | Telangiectasias | ATM | 11q22-q23 |
PNET, primitive neuroectodermal tumor; AML, acute myelogenous leukemia.
Neurofibromatosis 1
Neurofibromatosis 1 (NF-1), also known as von Recklinghausen disease, is the most common hereditary disease predisposing to CNS cancer, with a prevalence of 1 in 4000. The incidence is equal in men and women. It is an autosomal dominant disorder with 100% penetrance but is highly variable in its expressivity with both minimally and severely affected individuals within the same family. The NF-1 gene has been mapped to chromosome 17q11.2 and encodes a tumor suppressor, neurofibromin.
The predominant CNS tumors in NF-1 are optic pathway and brainstem gliomas. Optic pathway gliomas are usually pilocytic astrocytomas and brainstem gliomas are astrocytomas. The typical peripheral nerve tumor is the plexiform neurofibroma, which often involves the paraspinal and cranial nerves. Other NF-1 related tumors include malignant schwannomas, rhabdomyosarcomas, and GBMs.
Neurofibromatosis 2
Neurofibromatosis 2 (NF-2), known as central neurofibromatosis, is an autosomal dominant disorder that accounts for only 10% of all neurofibromatosis cases. The NF-2 gene is a tumor suppressor gene located on chromosome 22q12.2, which encodes a tumor suppressor, merlin. The predominant CNS tumors in NF-2 are vestibular schwannomas, often bilateral, and meningiomas.
Tuberous sclerosis
Tuberous sclerosis (TS) is an autosomal dominant disorder. It has been mapped to two different loci: TS complex 1 located on chromosome 9q34.14 encodes the protein hamartin and TS complex 2 located on chromosome 16p13.3 encodes the protein tuberin. The classic clinical triad of mental retardation, seizures, and facial angiofibromas occurs only in the most severe cases. Skin lesions are seen in 96% of patients, including angiofibromas, uncal fibromas, hypomelanotic skin patches known as “ash leaf spots,” and dental pits. The hallmark CNS tumor is the subependymal giant cell astrocytoma (SEGA). Although pathologically benign, SEGAs are driven by mTOR overactivity and recent work has demonstrated the value of mTOR inhibition with drugs such as everolimus causing tumor regression and improved seizure control.9
von Hippel–Lindau disease
von Hippel–Lindau (VHL) disease is a tumor syndrome involving multiple organ systems leading to hemangioblastomas in the cerebellum, spinal cord, and retina and pheochromocytomas and renal cell carcinoma. Other less common lesions include pancreatic and renal cysts and endolymphatic sac tumors. The hemangioblastomas are associated with overexpression of VEGF. VHL is an autosomal dominant disorder mapped to chromosome 3p25-p26. It has a high penetrance but variable expressivity.
Li–Fraumeni syndrome
Li–Fraumeni syndrome is a rare autosomal dominant disorder seen in children and young adults leading to multiple different tumors. It is caused by germ line mutations of p53. However, some families do not have the p53 mutation and their genetic defect is unknown. Overall penetrance of the gene is about 50% by age 30 and 90% by age 60. Common tumors include breast, osteosarcoma, and brain tumors, primarily high-grade gliomas. Some develop medulloblastomas and supratentorial primitive neuroectodermal tumors (PNET). Other less common tumors include soft tissue sarcomas, leukemia, and lung, adrenal, gastric, and colon cancers.
Brain tumor-polyposis/GI cancer syndromes
These hereditary syndromes are characterized by colon carcinoma with or without associated polyposis and malignant brain tumors. The two broad variants of brain tumor-polyposis syndrome vary in their association with malignant brain tumors: type 1 is associated with malignant gliomas and type 2 is associated with medulloblastoma. These syndromes incorporate autosomal dominant and autosomal recessive syndromes. The type 1 brain tumor-polyposis syndrome may not have polyposis and is due to mutations in one of the mismatch repair genes; these syndromes have also been called Turcot, Lynch, Muir-Torres, or Gardner syndrome. The type 2 brain tumor-polyposis syndrome is usually due to a germline mutation in the APC gene which has also been described as Turcot syndrome.
Histological classification of CNS tumors
WHO classifies tumors by their patterns of differentiation and presumed cell of origin. The majority of primary CNS tumors are neuroepithelial and the cell of origin is the glial cell (usually astrocyte) (Table 3).10 The remainder arise from meningeal, pituitary, lymphocytic, or germ cells. Neurons constitute less than 10% of brain cells and are an uncommon source of CNS neoplasms.
Table 3 Partial WHO list of common tumors of neuroepithelial tissue
| I. | Astrocytic tumors
|
| II. | Oligodendroglial tumors
|
| III. | Ependymal tumors
|
| IV. | Choroid plexus tumors
|
| V. | Neuronal and mixed neuronal-glial tumors
|
| VI. | Pineal tumors
|
| VII. | Embryonal tumors
|
In general, children most frequently develop PNETs, low-grade astrocytomas, and ependymomas; 70% are infratentorial and occur near the midline. In contrast, adults usually present with supratentorial tumors that are off the midline and are higher grade astrocytic tumors. GBM, a WHO grade 4 tumor, is hypercellular with nuclear pleomorphism, mitotic figures, endothelial proliferation, and necrosis. Gliosarcomas display the typical pathologic features of GBM in combination with densely packed spindle-shaped cells in herring bone patterns. Gliosarcomas have the same prognosis as GBM and are treated identically. Anaplastic astrocytomas (AA), WHO grade 3 tumors, also have increased cellularity, nuclear atypia, and mitoses, but necrosis and microvascular proliferation are absent. WHO grade 2 astrocytomas typically comprise a fairly uniform group of astrocytes in a background of fibrillary matrix. There is less cellular pleomorphism, and mitoses are rare; there is an absence of vascular proliferation and necrosis. Grade 2 and 3 tumors with an IDH mutation have a better prognosis than tumors matched for type and grade without an IDH mutation.
Glial tumors can be very heterogeneous; there can be different grades and even different histologic features with astrocytic and oligodendroglial appearing regions within the same tumor. This can be challenging diagnostically and highlights the need to get an adequate pathologic sample for examination. The highest grade found within the lesion determines the diagnosis and dictates treatment. Grade 1 tumors, juvenile pilocytic astrocytomas (JPA), occur almost exclusively in children. They are characterized by low cellularity, Rosenthal fibers, and cysts, and they do not infiltrate surrounding tissue extensively. They may contain rare mitoses and hyperchromatic nuclei, although these are not features of malignancy in these tumors. In young children, they occur most commonly in the cerebellar hemispheres where they have a 95% 5-year progression-free survival (PFS) rate after complete resection. The other low-grade glial tumors also tend to be discreet and less infiltrative. These include pleomorphic xanthoastrocytoma (PXA), ganglioglioma, neurocytoma, and dysembryoplastic neuroepithelial tumor (DNET). These tumors usually require an experienced neuropathologist to diagnose.
Oligodendrogliomas are characterized by a relatively uniform array of small round cells with artifactual perinuclear halos in a background of fine capillary (“chicken-wire”) vasculature. They are typically described as having a “fried-egg” appearance. Occasionally, oligodendrogliomas have features of anaplasia, pleomorphism, and necrosis and are classified as grade 3 anaplastic oligodendrogliomas (AO). They have a better clinical prognosis than their astrocytic counterparts. Some oligodendrogliomas have loss of heterozygosity on chromosomes 1p and 19q due to a translocation. Tumors with 1p19q codeletion have a better prognosis and are more responsive to therapy. Tumors containing a mutation in IDH and have 1p19q codeletion have the best prognosis.
PNETs are a group of neoplasms characterized by undifferentiated small blue cells with Homer–Wright rosettes (cells arranged around a true lumen). They are usually fast growing and often disseminate along CSF pathways. They are most common in children and are often found in the cerebellum (medulloblastoma) and in the pineal gland (pinealoblastoma). Although these lesions are histologically identical, the medulloblastoma is more amenable to treatment and has a better outcome.
Ependymomas arise from ependymal cells that line the ventricles and spinal canal. They arise wherever ependymal cells are present and have a predilection for the fourth ventricle. They are the most frequent neuroepithelial tumor of the spinal cord, accounting for more than 50% of spinal gliomas in both children and adults. They are histologically characterized by perivascular pseudorosettes (tumor cells arranged radially around blood vessels) and Homer–Wright rosettes. These tumors are usually low grade but can be aggressive and spread along CSF pathways.
Clinical presentation
Patients with brain tumors may present with generalized, nonfocal symptoms and signs or with focal manifestations related to the specific location of the tumor in the brain.10 Factors that contribute to the presenting symptoms and signs include tumor location, size, and growth rate. Supratentorial tumors are more likely to present with seizure, whereas infratentorial tumors more commonly present with headache, nausea, and vomiting. Superficial cortical tumors are more likely to cause seizure, whereas deeper tumors are more likely to cause personality and cognitive changes. Seizure is the presenting symptom in about 20% of patients with malignant brain tumors, but is the initial manifestation in approximately 90% of those with low-grade tumors. Tumors in eloquent areas of the brain will cause focal symptoms such as aphasia, hemiparesis, or sensory loss. Tumors in the brainstem typically present with cranial nerve deficits such as diplopia or facial weakness. Tumors of the cerebellum may cause ipsilateral ataxia, unsteady gait, and nystagmus.
Symptoms usually evolve over several weeks; sometimes the symptoms begin acutely, corresponding to a hemorrhage into a metastasis or glial tumor, or a seizure with a prolonged postictal state. GBM and oligodendrogliomas of any grade have a 5–10% risk of hemorrhage. Any intracranial metastatic tumor can hemorrhage, but certain tumors, such as melanoma, thyroid, renal, and choriocarcinoma, have a propensity to bleed; however, lung cancer is the most common source of a hemorrhagic brain metastasis because of its high frequency of CNS metastases. On occasion, focal seizures can cause neurologic signs that do not resolve completely.
Diagnostic neuroimaging
Patients with symptoms and signs suggestive of an intracranial lesion should have a magnetic resonance imaging (MRI) study; it is the modality of choice for CNS tumors and CT should be used only in those patients unable to get an MRI.
High-grade tumors such as GBM disrupt the blood–brain barrier and have the characteristic appearance of a hypointense center surrounded by a hyperintense irregular rim of contrast enhancement (Figure 1). Almost 95% of GBMs will enhance with gadolinium. Anaplastic gliomas enhance less frequently, particularly in younger adults, and they may be confused with lower grade gliomas radiographically (Figure 2). GBMs also have a greater tendency to spread along white matter tracts, particularly the corpus callosum, and to cross to the contralateral cerebral hemisphere. In comparison, low-grade tumors have an intact blood–brain barrier and usually do not contrast enhance. An exception is the JPA, a grade 1 tumor that has areas of dense enhancement. Fluid-attenuated inversion recovery (FLAIR) sequences provide rapid distinction between normal brain and brain tumor or edema and provide the best images to delineate the radiographic extent of the lesion (Figure 3). On FLAIR imaging, however, infiltrative tumor cannot be differentiated from edema. Diffusion-weighted imaging (DWI) assesses the mobility of water molecules and may differentiate between ischemia and tumor. Perfusion images measure blood volume and vascularity and correlate with tumor grade and with 18fluorodeoxyglucose (FDG) positron emission tomography (PET) studies.
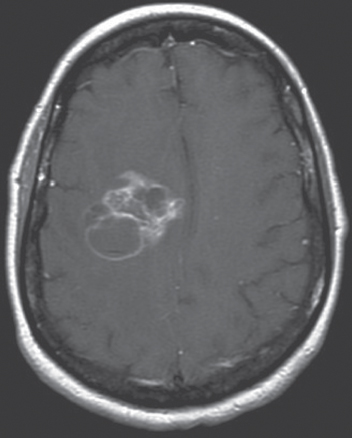
Figure 1 Glioblastoma. This is a postgadolinium T1-weighted MRI of a right posterior frontal glioblastoma. There is irregular contrast enhancement with focal areas of necrosis.
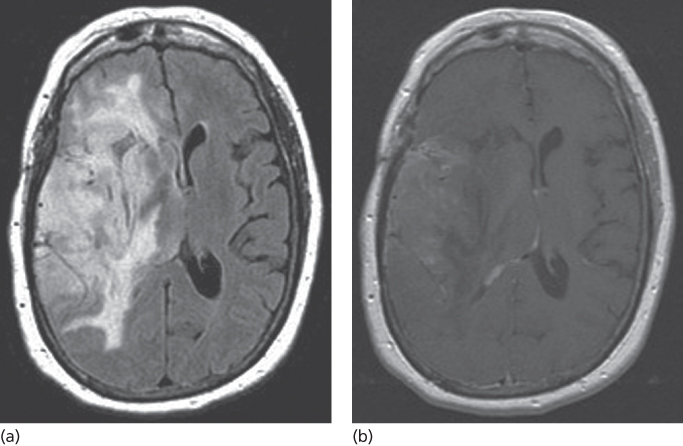
Figure 2 Anaplastic oligodendroglioma. (a) FLAIR image showing extensive tumor infiltrating the right hemisphere. (b) Patchy enhancement is seen throughout the tumor.
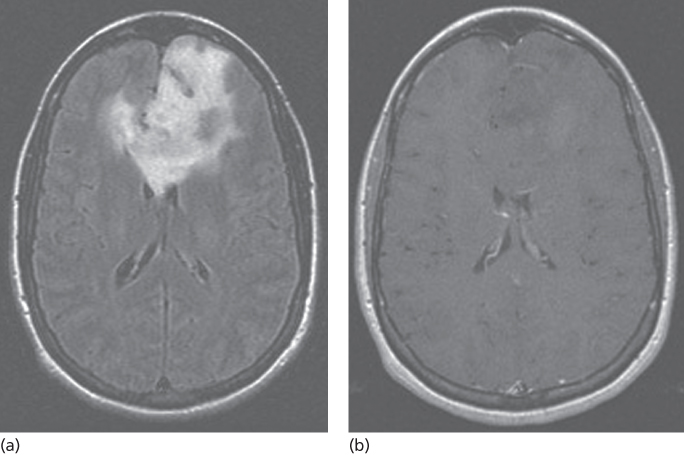
Figure 3 Low-grade glioma. (a) FLAIR image demonstrating extensive infiltrative disease predominantly of the left frontal lobe extending across the anterior corpus callosum and involving the deep right frontal white matter. (b) Postcontrast images show no evidence of enhancement of this large lesion.
On MRI, metastases are usually spherical and have more regular margins than primary tumors. They are usually found at gray–white matter junctions in watershed areas of the brain. When small, they uniformly contrast enhance, and when larger, they may ring enhance. The tumor is usually surrounded by substantial edema. Very small metastases may appear as small dots of contrast enhancement with or without hyperintensity on the FLAIR image; they usually lack surrounding edema. Fifty percent of patients have a single identifiable brain metastasis, 20% have two metastases, 13% have three and the remainder have more than three. There are some limitations to MRI, and the differential diagnosis of a brain tumor includes radiation necrosis, ischemic stroke, infection, inflammatory process, and demyelination.
Other imaging techniques
PET has several uses in the diagnosis of brain tumors. PET can help distinguish between recurrent tumor and radiation necrosis, may differentiate low-grade from high-grade lesions, and may guide stereotactic biopsy to the site of high-grade tumor within an apparently low-grade lesion seen on MRI (Figure 4). PET is performed by injecting substances such as glucose, an amino acid such as methionine, or even a nucleotide labeled with a positron-emitting isotope such as O15, C11, N13, and F18. FDG is the most commonly used isotope for evaluating brain tumors, and defines the metabolic rate of the lesion being examined. Hypermetabolism (increased FDG uptake) is common in high-grade tumors and hypometabolism (low FDG uptake) is common in low-grade tumors. FDG PET may help distinguish radiation necrosis from recurrent tumor. Whereas both may appear similar on MRI, radiation necrosis is typically, but not always, hypometabolic on PET in comparison to recurrent high-grade tumor, which is either isometabolic with normal brain or hypermetabolic on PET.
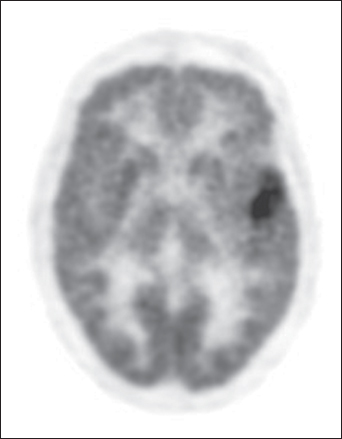
Figure 4 Low-grade glioma. This is an FDG PET image of a left temporal low-grade glioma showing an area of focal hypermetabolism that corresponded to a focal area of anaplasia.
Functional magnetic resonance imaging (fMRI) maps the functional organization of the brain, particularly the primary motor, sensory, and language cortices. fMRI is based on the concept that increased neuronal activity results in increased cerebral blood flow (CBF), causing a focal change in the oxyhemoglobin to deoxyhemoglobin ratio leading to increased signal on MRI. The fMRI is useful in presurgical planning and allows a surgeon to map eloquent cortex and then plan resection while preserving neurologic function; this facilitates maximal resection without neurologic deficit.
Principles of therapy
Treatment of a brain tumor includes both definitive and supportive therapy. Definitive therapy encompasses surgery, radiation therapy, and chemotherapy. Supportive therapy considers management of tumor symptoms such as treatment of focal and general symptoms with corticosteroids, seizure control with antiepileptic medication, treatment of deep venous thrombosis (DVT) with anticoagulants, and the provision of psychosocial support when needed.
Supportive therapy
In addition to surgery, RT, and chemotherapy, where the goal of treatment is to extend survival, various types of supportive therapy are also required to treat a patient’s neurologic symptoms and improve their quality of life.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


