INTRODUCTION
SUMMARY
Polycythemia vera (PV) is classified in the group of Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) that also includes essential thrombocythemia (ET) and primary myelofibrosis (PMF). Chronic myelogenous leukemia was historically classified as a MPN, but is now considered a separate entity. PV is an acquired primary clonal polycythemic disorder. Primary polycythemias result from abnormal intrinsic properties of erythroid progenitors that proliferate independently or excessively in response to extrinsic regulators; low serum erythropoietin is their hallmark. PV is the most common primary polycythemia. It arises from mutation(s) of a pluripotent hematopoietic stem cell, which results in excess production of erythrocytes and variable overproduction of granulocytes and platelets. It is often accompanied by splenomegaly. Most patients with PV have a somatic mutation of the Janus-type tyrosine kinase-2 gene (JAK2) that is detectable in blood myeloid cells. The mutation results in constitutive hyperactivity of JAK2 kinase stemming from loss-of-function of its negative regulatory domain. The most common mutation is JAK2V617F, which is present in virtually all cases of PV; a small minority of PV patients have a mutation in other parts of JAK2 (exon 12). The JAK2V617F mutation is also found in many patients with ET and myelofibrosis (MF), albeit at lower frequency (55 percent in ET and 65 percent in MF). As with other clonal hematologic disorders, PV can undergo a clonal evolution to PMF (JAK2V617F-positive) or acute leukemia (either JAK2V617F-negative or positive). In virtually all JAK2V617F-positive PV patients, at least some progenitors exist that become homozygous for the JAK2V617F mutation by uniparental disomy-acquired mitotic recombination. The majority of these progenitors account for the erythropoietin-independent erythroid colonies detected in vitro by clonogenic burst-forming unit–erythroid assay. The JAK2V617F mutation is often not the initial cause of clonal proliferation, but may be preceded by other germline and somatic mutation(s) (e.g., TET2).
Arterial and venous thromboses are a major cause of morbidity and mortality in PV, and a small proportion of patients develop secondary myelofibrosis (sometimes called the spent phase) and/or an invariably fatal acute leukemic transformation. Myelosuppressive therapy has been an effective mode of therapy, with drugs such as hydroxyurea, busulfan, pipobroman, and radioactive phosphorus useful in controlling proliferation of all blood cell lineages. However, while myelosuppressive therapy controls the cellular proliferation and decreases the incidence of thrombotic complications, many of these drugs have leukemogenic potential. In contrast, pegylated interferon-α may lead to complete hematologic remission and restoration of polyclonal hematopoiesis and avoid the leukemogenic complications. Targeted therapy with JAK2 kinase inhibitors is currently being evaluated in clinical trials and, thus far, have been found effective in decreasing the need for phlebotomies, decreasing the number of white cells and splenomegaly, and improving the patient’s quality of life.
Acronyms and Abbreviations
AML, acute myelogenous leukemia; BFU-E, burst-forming unit–erythroid; ECLAP, European Collaboration on Low-Dose Aspirin in Polycythemia Vera; EEC, endogenous erythroid colony; ELN, European Leukemia Net; ET, essential thrombocytosis; HDAC, histone deacetylase; HU, hydroxyurea; IFN-α, interferon-α; IWG-MRT, International Working Group for Myeloproliferative Neoplasms Research and Treatment; JAK2, Janus-type tyrosine kinase 2; MDS, myelodysplastic syndrome; MF, myelofibrosis; MPN, myeloproliferative neoplasm; PCR, polymerase chain reaction; PEG-IFN, pegylated interferon; PFCP, primary familial and congenital polycythemia; PMF, primary myelofibrosis; PV, polycythemia vera; SNP, single nucleotide polymorphism; UPD, uniparental disomy; TET2, a homologue of chromosome 10-11 translocation; WHO, World Health Organization.
DEFINITION AND HISTORY
The term polycythemia, denoting an increased amount of blood, has traditionally been applied to those conditions in which the mass of erythrocytes is increased. In polycythemia vera (PV), an increase in the erythroid mass is frequently accompanied by an increase in neutrophils and platelets. For a classification of the polycythemias, see Chap. 57 and Chap. 34, Table 34–2. Although several clinical stages of PV are recognized (masked PV, plethoric phase, stable phase, transformation, spent phase, and acute leukemia), it is not clear whether these stages represent a sequential progression of the disease or whether all patients progress through all stages.
PV, the sole clonal form of primary polycythemia, was first described in 1892 by Vaquez.1 In 1903, Osler reviewed four of his own PV cases and an additional five cases from the literature and wrote, “The condition is characterized by chronic cyanosis, polycythemia, and variable moderate enlargement of the spleen. The chief symptoms have been weakness, prostration, constipation, headache, and vertigo.”2 The increased proliferation of granulocyte precursors and megakaryocytes was first described by Türk in 1904.3
EPIDEMIOLOGY
A recent analysis of 20 studies of PV patients from around the world revealed an annual incidence rate of 0.84 cases per 100,000 people, with no bias for gender.4,5 The true incidence may be higher, as many cases are asymptomatic and thus not diagnosed. Testing for the Janus-type tyrosine kinase 2 (JAK2)V617F mutation can uncover hidden cases of PV among subjects with thrombosis or concomitant iron deficiency. The incidence of PV may be higher among Ashkenazi Jews.6,7
Although most patients with PV do not have a history of polycythemia in the family, familial incidence of the disorder is known to occur8,9,10 and is very likely underreported. In a large Swedish study of more than 25,000 first-degree relatives of 11,000 myeloproliferative neoplasm (MPN) patients, the incidence of MPNs were five to seven times higher in relatives than in controls.11 In familial cases of PV, an inherited predisposition, perhaps in the form of a germline mutation, presumably facilitates the acquired somatic mutation(s) necessary for disease onset.9,12
ETIOLOGY AND PATHOGENESIS
PV arises from the neoplastic transformation of a single normal hematopoietic pluripotent cell, which acquires a selective proliferative and survival advantage, resulting in the development of a variable degree of clonal hematopoiesis. Once large enough, the clone then suppresses and replaces normal polyclonal hematopoiesis. The clonal origin of PV has been demonstrated in women heterozygous for a polymorphic X-chromosome marker such as, glucose-6-phosphate dehydrogenase13 as well as by more modern clonality assays (Chap. 10).14 In both cases, all hematopoietic cell lineages9 express either one isoform of the enzyme, or some polymorphic allele encoded by the maternal or paternal X chromosome, whereas T lymphocytes and nonhematopoietic cells are a mosaic of both enzyme types.
In vitro marrow- or blood-derived erythroid colonies of PV patients arise from both normal burst-forming unit–erythroid (BFU-E) precursors and BFU-E precursors that are erythropoietin-independent. Erythropoietin-independent BFU-E precursors form so-called endogenous erythroid colonies (EECs),15,16,17,18 a characteristic feature of PV. The fibroblasts that accumulate in the marrow of patients with PV as the disease progresses are not part of the abnormal PV clone. Rather, they seem to accumulate in response to cytokines released by megakaryocytes and other cells (Chap. 86).19
Other abnormalities that have been described include decreased levels of a platelet thrombopoietin receptor,20 deregulation of bcl-x, an inhibitor of apoptosis,21 increased expression of protein tyrosine phosphatase activity by red cell precursors,22 and acquired loss-of-heterozygosity of chromosome 9p as a result of uniparental disomy (UPD).9 This last observation was one of two routes that led to the discovery of the JAK2 2343G > T mutation encoding the V617F mutation located on chromosome 9p,12,23 which has improved our understanding of disease pathogenesis, improved the specificity of diagnosis, and led to an explosion of research in MPN (see “JAK2V617F Mutation” below).
There are no specific karyotypic markers occurring with high frequency in PV. Fewer than 25 percent of patients have karyotypic abnormalities at diagnosis,24,25,26,27,28,29 but the incidence rises with the increasing duration of the disease,25,30 suggesting that karyotypic abnormalities represent secondary genetic events.31 Cytogenetic abnormalities may potentially herald transformation from PV to myelofibrosis, acute myeloid leukemia, or a myelodysplastic syndrome, but as of now, these associations are weak.29
JAK2 kinase is present in all hematopoietic cells and is essential for proliferative intracellular signaling in response to a variety of hematopoietic growth factors (Chaps. 34 and 57). The V617F mutation was first identified in PV in 2004,23 and was simultaneously reported by several laboratories.32,33,34 The V617F mutation is present in virtually all patients with PV and in more than 50 percent of patients with essential thrombocytosis (ET; Chap. 85) and myelofibrosis (MF; Chap. 86); rarely is it found in the minority of patients with other myeloproliferative disorders.35,36 In PV (unlike in ET), it is often in its associated homozygous form as a result of UPD, at least in some of the progenitors.26,37 Patients bearing homozygous JAK2V617F tend to have a longer duration of disease,33 higher hemoglobin levels, and increased incidence of pruritus38 and are more likely to transform to post-PV MF (Chap. 86).39 The JAK2V617F allele burden in PV may also be correlated with increased spleen volume, increased leukocytosis, and severity of MF.39,40,41,42 It should be noted, however, that PV patients can achieve a complete hematologic remission without a significant molecular response (i.e., a decrease in JAK2V617F allele burden).43 In some of the rare PV patients who are JAK2V617F-negative, a different JAK2 mutation is present in exon 12.44 Several different JAK2 exon 12 mutations, including missense mutations, insertions, and deletions, have been described. Patients with exon 12 mutations may present with different clinical manifestations from those with the classic JAK2V617F mutation: erythrocytosis only, higher hemoglobin, and differences in marrow morphology.45 Disease course and clinical outcome, however, are similar.46
Studies of families of MPN patients, in which several different MPNs occur in a single pedigree,47 indicate that JAK2 mutations may not be solely responsible for the disease phenotype and may not even represent the disease-initiating event. A number of compelling lines of evidence support this conclusion. First, in familial PV, there is no clear linkage between the disease and chromosome 9p, the genetic site of JAK2, suggesting an independent germline predisposition to PV.12 Second, in familial PV, affected members can be either JAK2V617F– positive or negative.48 Third, acquisition of the JAK2V617F mutation may be a late genetic event.49 Fourth, in sporadic PV, only a proportion of clonal PV cells are JAK2V617F-positive.37 And fifth, acute leukemic transformation of any JAK2-positive MPN, including PV, is frequently negative for the JAK2V617F mutation.35,50 These diverse observations strongly suggest that the somatic mutation of the JAK2 gene is not the initiating or sole pathogenic process in PV, but in most patients is essential for the clinical phenotype of PV. The pathways leading to acquisition of the JAK2V617F mutation, homozygosity of JAK2V617F, and participation of many other genes in the 9p UPD region may have phenotypic and prognostic significance.51,52 Additional prognostic significance can be ascertained by analysis for clustering of specific genes.53
A genomic chromosome 9p functional variant might also be relevant to the pathogenesis of JAK2V617F. Independent occurrence of the JAK2V617F mutation on different haplotypes was found, although a specific constitutional inherited JAK2 haplotype (GGCC, 46/1), associated with the JAK2V617F somatic mutation, was found in most JAK2V617F-positive individuals.54 The risk of acquiring a JAK2V617F-positive MPN is three to four fold higher in patients with the JAK2 GGCC (46/1) haplotype.54,55,56,57 This GGCC haplotype of JAK2 also confers susceptibility to JAK2 exon 12 mutation-positive PV.55 These studies suggest that pre-JAK2 hypermutability events exist and that germline genetics play an important role in the early pathogenesis of MPNs.
In addition to the important role of JAK2V617F and other JAK2 mutations in the etiology of PV and other MPNs, mutations in other genes may be important to the full genesis of these disorders.
TET2 is a homologue of the gene originally discovered at the chromosome ten-eleven translocation (TET) site in a subset of patients with acute leukemia. TET2 mutations were found in hematopoietic cells from a significant proportion of patients with PV and other MPNs.58,59 It has been established that TET2 loss-of-function mutations originate in pluripotent hematopoietic stem cells but seem to favor myeloid rather than lymphoid proliferation, and that in many patients both TET2 alleles were affected. However, studies in familial PV demonstrated that the TET2 mutation is often not disease-initiating, as the TET2 mutations differ among affected relatives and, in some instances, the TET2 mutations followed, rather than preceded, the appearance of JAK2V617F.60 Additionally, recurrent TET2 mutations have been reported in elderly patients with clonal hematopoiesis but no evidence of hematological malignancy.61 Several additional genes commonly bear mutations in PV patients, including ASXL1, DNMT3A, and IDH1/2.62,63 The quantitative proportion of clones carrying different mutations may change during disease progression.64
Although JAK2 kinase is clearly involved in the pathogenesis of PV, immune dysfunction and chronic inflammation may also be implicated. A history of any autoimmune disorder is associated with a 20 percent increased risk of developing an MPN,65 and chronic inflammation is suggested to contribute to mutagenesis and clonal evolution in PV.66,67,68 A recent molecular profiling study found that a number of immune and inflammatory genes were either up- or downregulated in PV patients, among them interleukin-10, interleukin-4, complement 5, short pentraxin C-reactive protein, fibrinogen, orosomucoid, and transforming growth factor-β1.69 The dysregulation of immune and inflammatory genes in PV may represent an additional avenue for future therapeutic development.
CLINICAL FEATURES
PV usually has an insidious onset, most commonly during the sixth decade of life, although onset may occur from childhood to old age.70 Presenting signs and symptoms may include headache, plethora, pruritus, thrombosis, and gastrointestinal bleeding, but many patients are diagnosed because elevated hemoglobin, and sometimes other cell counts, are found on a periodic medical examination. Others cases may be uncovered during investigation for blood loss, iron-deficiency anemia, or thrombosis. Symptoms are reported by at least 30 percent of patients at the time of diagnosis; other patients may admit to symptoms on direct questioning. The most common symptoms, in decreasing order of frequency, are headache, fatigue, weakness, pruritus, dizziness, and night sweats, but these symptoms are more likely in those PV patients transforming to MF (Chap. 86).70
PV generally occurs in older patients, a population who may already have an elevated rate of vascular abnormalities (e.g., coronary artery disease). Development of PV represents an additional increase in the risk of vascular events.
Thrombotic episodes are the most common and most important complication of PV,71,72,73 occurring in approximately one-third of PV patients.74 From one-half to three-quarters of these events are arterial75; ischemic strokes and transient ischemic attacks account for the majority of arterial complications. In some studies, 40 to 60 percent of patients develop at least one thrombotic event over a period of 10 years, the annual incidence being approximately equal throughout this period.76 However, in prospective studies, thrombosis was most common just prior to and during the first few years after diagnosis.74,77,78 The most common serious complication is a cerebrovascular accident, which accounts for approximately one-third of thrombotic events, followed in frequency by myocardial infarction, deep vein thrombosis, and pulmonary embolism.76 The allelic burden of the JAK2V617F mutation has been correlated with activation of thrombotic pathways in PV patients,79,80 although this idea is not universally accepted.39
Bleeding and bruising is a common complication of PV, occurring in approximately one-quarter of patients in some series.74 Although such episodes are usually minor (e.g., gingival bleeding, nose bleeding, easy bruising), serious gastrointestinal and other hemorrhagic complications with a fatal outcome can also occur.31,75,81,82
Splanchnic Vein Thrombosis Splanchnic vein thrombosis, including hepatic veins, portal vein or a mesenteric vein may occur. Hepatic vein thrombosis (Budd-Chiari syndrome) is the most common site of involvement. Budd-Chiari syndrome is a catastrophic and often fatal complication of PV. In one series, it occurred in 10 percent of 140 PV patients,83 but was less common in a European collaborative study.75 Budd-Chiari syndrome is caused by a thrombosis in the hepatic venous outflow, leading to ischemia from reduced perfusion through hepatic arterioles and necrosis of hepatocytes. Budd-Chiari syndrome may present as ascites with or without right-upper-quadrant abdominal pain, hepatosplenomegaly, and jaundice.
Budd-Chiari syndrome may be the first clinical manifestation of PV; endogenous erythroid colony formation and JAK2V617F mutation have been described in many of these patients before any clinical evidence of polycythemia.84,85 PV is the most frequent underlying disease associated with Budd-Chiari syndrome. The association of Budd-Chiari syndrome and PV is so strong that many experts advocate screening for PV with JAK2V617F mutation analysis in all patients who present with hepatic vein thrombosis.86,87 Budd-Chiari syndrome is a serious condition, often requiring a liver transplant for treatment.85,88,89
Pruritus occurs in approximately 40 percent of PV patients.90 It is usually aggravated by bathing or showering (aquagenic pruritus), and may be so severe it markedly compromises the quality of life of the patient.82,91 It has been attributed to increased numbers of mast cells in the skin92 and to elevated histamine levels,93 although these associations were not found in other studies.94
Several PV patients have developed the dermatologic disorder, acute febrile neutrophilic dermatosis (i.e., Sweet syndrome).95,96
Erythromelalgia is a syndrome characterized by warmth of the extremities; painful, reddened digits; a burning sensation; and erythema of the fingers, hands, and feet (Fig. 84–1) that is associated with thrombocytosis. It characteristically responds rapidly to low-dose aspirin therapy. In severe cases, it results in ischemic necrosis of the digits and may lead to their amputation. This syndrome occurs in less than 5 percent of PV patients.75,81 It is not specific to PV or other MPNs, and in one series of 168 patients with erythromelalgia, less than 10 percent had PV.97 A causative role for transient microvascular occlusion by platelet aggregates has been proposed (Chap. 112).98,99
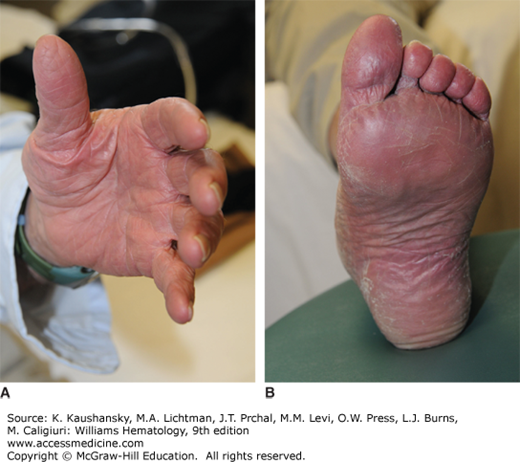
Portal hypertension, varices, and abdominal pain are not uncommon,100 and are often caused by cryptic or overt thrombosis in the hepatic, portal, or mesenteric veins. The former site (Budd-Chiari syndrome) is most common. The occurrence of Budd-Chiari syndrome is noted above (See Splanchnic Vein Thrombosis above). The incidence of peptic ulcer is four to five times greater than in the general population.101 Gastrointestinal bleeding may be the first presenting symptom of PV, with iron deficiency caused by gastrointestinal blood loss frequently masking erythrocytosis.87
Cardiovascular complications include angina, myocardial infarction, and congestive heart failure, related to a predisposition to thrombosis in the coronary circulation.31,75,77
Pulmonary hypertension occurs in a higher than expected frequency in patients with PV. The suggested etiologies include smooth muscle hyperplasia induced by the release of platelet-derived growth factor from activated platelets, obstruction of pulmonary circulation by megakaryocytes, extramedullary hematopoiesis, and unrecognized recurrent thrombotic events.102,103 None of these etiologies are clearly established.
Neurologic symptoms such as dizziness and headache are very common in PV.31,75,77,81,104 Spinal cord compression secondary to extramedullary hematopoiesis has been documented.105
The increased nucleic acid turnover that results from the excessive proliferation of marrow cells often leads to an increase in blood uric acid concentration; gout can be exacerbated in some patients.31
More than 75 percent of patients with uncontrolled PV develop complications during or after major surgery because both bleeding and thromboses are common.82,106 Thus, it is advised to normalize blood counts and blood volume before surgical interventions, which may lower the frequency of intraoperative and postoperative complications.
Chapter 8 discusses the complications of PV in pregnancy.
The spent phase of PV, also referred to as post-PV MF, is a frequent and often terminal complication of the disease.75,78,107 It is characterized by a combination of anemia (non–iron deficiency), progressive increase of splenic size (Fig. 84–2), and marrow fibrosis (Chap. 86). The spent phase may first be noticed when phlebotomy requirements decrease. Thrombocytosis and granulocytosis (often with immature myeloid cells) are common. In a minority of cases, thrombocytopenia and granulocytopenia may occur. Affected individuals are frequently symptomatic with anemia, bleeding, splenic enlargement with early satiety, and/or upper abdominal pain secondary to splenic infarcts. Most patients become dependent on transfusions or erythropoietin therapy.31,75,77,81,107 Development of the spent phase is associated with an increased risk of leukemic transformation31,108; in the PV Study Group-01 study, the incidence of acute leukemia was 24 percent in PV patients with MF, compared to 7 percent in those without MF, although it should be noted that a sizable number of the patients in this study had been treated with alkylating agents or 32P.78 Therapy, including use of JAK2 inhibitors, is described in Chap. 86.
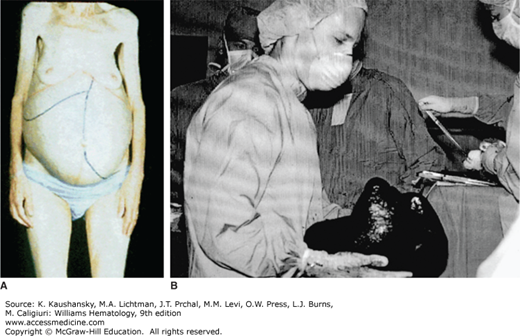
Patients with PV have an increased risk of developing leukemia.71,82 This contrasts with other nonclonal polycythemic disorders, wherein progression to leukemia is not part of the disease process. Acute leukemia, usually myelogenous, is an invariably fatal complication of PV. A European multicenter observational study of 1638 patients reported a 6.3 percent relative risk of developing leukemia within 10 years after the diagnosis of PV.75 Different treatments can also influence the risk of transformation from PV to leukemia. In the PV Study Group-01 randomized trial, the incidence of acute leukemia at 18 years of followup was 1.5 percent on the phlebotomy-only treatment arm, 10 percent on the 32P treatment arm, and 13 percent on the chlorambucil treatment arm.81 In a more recent study of 1638 patients, the risk of transformation to acute myelogenous leukemia (AML) or a myelodysplastic syndrome (MDS) in PV patients also varied by treatment. Treatment-specific risk of transformation, ordered by increasing risk, was: phlebotomy (hazard ratio [HR]: 0.91), hydroxyurea (HR: 1.09), interferon (INF)-α (HR: 1.24), busulfan (HR: 8.64), pipobroman (HR: 4.32), and 32P (HR: 8.96).109 Leukemic transformation was not significantly different between phlebotomy, hydroxyurea, and INF-α treatments.109
Acute leukemia as the terminal PV event may arise from either the JAK2V617F-positive clone or, more frequently, from a hematopoietic cell that does not carry the JAK2 mutation.35,47,50
LABORATORY FEATURES
The hemoglobin concentration, erythrocyte count, and hematocrit are usually increased, and the mean cell volume is usually low-normal or low in untreated patients, and low in patients who have undergone phlebotomies or had gastrointestinal bleeding episodes. Red cells are hypochromic and microcytic, with morphology characteristic of iron deficiency. Although increased hemoglobin is generally a diagnostic feature of PV, at times the hemoglobin level may be low, normal, or borderline, a condition termed “masked” PV.110,111,112
The appearance of significant aniso- and poikilocytosis and teardrop cells (dacryocytes; Chap. 31) heralds the onset of the spent phase and post-PV MF (Chap. 86).
The Polycythemia Vera Study Group employed the direct determination of red cell mass as the sine qua non of the diagnosis of PV for all patients entered into their studies.76 Some believe that even in the routine clinical setting, this procedure should be performed on all patients to establish a diagnosis of PV.76,113 Unfortunately, the determination of red cell mass is expensive, requires the use of radioactive isotopes in patients, and, when performed by the inexperienced, is often inaccurate.114 Red cell mass determination is not useful in distinguishing PV from secondary polycythemia because the red cell mass is increased in both disorders. The principal value of red cell mass determination is to distinguish apparent or spurious polycythemia from PV and secondary polycythemia.110,111 It could also be useful in distinguishing cases of masked PV from ET.115 The availability of the JAK2 assay has made red cell mass determination only rarely important.
Absolute neutrophilia occurs in approximately two-thirds of PV patients.70 Occasional myelocytes and metamyelocytes are present in the blood, and considerable degrees of cell immaturity are present in patients with longstanding, advanced disease. Again, these abnormalities herald the onset of the spent phase (Chap. 86). Basophilia occurs in approximately two-thirds of patients with uncontrolled disease.31,81,116 In PV, the proportion of activated neutrophils is increased,117 and it is possible that neutrophils may be an important factor in PV-associated thrombosis.118
The leukocyte alkaline phosphatase level is elevated in approximately 70 percent of patients with PV,70 but this assay has now become largely obsolete.
The platelet count is increased in approximately 50 percent of PV patients at the time of diagnosis, and in approximately 10 percent of patients it is greater than 1000 × 109/L.70 There are no consistent abnormalities of thrombopoietin levels in PV patients.119 A significant proportion of PV patients first present with isolated thrombocytosis without elevated hemoglobin, especially if the patient has a reason for iron deficiency, and are sometimes misdiagnosed with essential thrombocythemia.120
Qualitative platelet abnormalities have been described which may play a role in the pathogenesis of thrombotic and hemorrhagic events. In vitro spontaneous platelet aggregation is accelerated. On the other hand, patients with MPNs often display a nearly pathognomonic defect in the primary wave of platelet aggregation induced by epinephrine (Chaps. 112 and 121).121
There are a number of additional platelet abnormalities associated with PV, including increased platelet thromboxane A2 generation122 and increased excretion of thromboxane metabolites.123 Platelet factor-4 levels are elevated.124 Platelet survival may be shortened.124,125 Fibrinogen binding after stimulation with a platelet-activating factor is diminished,126 and there is reduced expression of the thrombopoietin receptor.20 However, none of these changes are specific for PV. In a prospective study, a PIA2 polymorphism of the platelet glycoprotein IIIa was associated with an increased risk of arterial thrombosis in PV patients,127 although this conclusion remains controversial.
Platelet counts greater than 1000 to 1500 × 109/L are associated with progressive decrease of von Willebrand factor (an acquired, type 2 von Willebrand disease) and increased risk of bleeding, but not of thrombosis.128
Serum lysozyme levels are slightly increased in some patients,129 and because of increased leukocyte turnover and increased levels of B12 binding protein, serum B12 determination is usually increased.130 Hyperuricemia, a consequence of hyperproliferative myelopoiesis, is frequently encountered.31
Mutations within the JAK2 gene cause deregulation of the hematopoietic process, which is expressed in a wide spectrum of disorders involving the expansion of erythrocytes, granulocytes, or leukocytes. The primary lesion associated with these disorders was discovered in exon 14: A single nucleotide change JAK2 G1849T resulting in the amino acid substitution V617F. Detection of the JAK2V617F mutation provides a qualitative diagnostic marker for the identification of the Philadelphia-chromosome-negative subgroup of MPNs, and differentiates them from congenital and acquired reactive hematopoietic disorders. In general, the JAK2V617F allele burden is lower in ET patients than in either PV or primary myelofibrosis (PMF).131,132 In many, but not all, JAK2V617F-positive PV patients, at least some progenitors exist that became homozygous for the JAK2V617F mutation by UPD acquired by mitotic recombination.37,133
Allele-specific polymerase chain reaction (PCR) is widely used for single nucleotide polymorphism (SNP) genotyping; the technique is based on amplification of DNA by an allele-specific primer matching the polymorphism at the 3′ position. This approach is directly applicable to analysis of JAK2V617F because the mutation (G1849T) is analogous to a SNP. To improve the specificity, sensitivity, and reliability for quantitating the JAK2V617F allele burden, two modifications of technique were incorporated: Inclusion of a second mismatch at the –1 position, and substitution of a modified locked nucleic acid at the –2 position. A study comparing 11 different techniques was undertaken and carried out in 16 laboratories using various testing platforms.134 Although five of the 11 techniques were similarly reliable for quantification of JAK2V617F loads that were equal to or greater than1 percent of total JAK2V617F, the allele-specific quantitative PCR technique could detect 0.2 percent of JAK2V617F.
The majority of laboratories analyze quantitative assays of JAK2V617F allele burden from (clonal) granulocytes; nonquantitative analyses use total leukocytes, whole blood, or marrow for screening. A proportion of JAK2V617F-negative assays are positive using sensitive quantitative analyses.134 Plasma has been used for detection of the JAK2V617F DNA and mRNA mutation and zygosity state,135,136 but plasma analysis is not reliable.137
Although the most common JAK2 mutation is a single SNP in exon 14, many MPN cases negative for the exon 14 JAK2V617F mutation may carry one of a number of mutations in exon 12. Almost 40 different mutations in exon 12 have been identified within codons 536 to 547, including substitutions, deletions, and duplications.45,138,139,140,141,142 In addition to the various mutations observed in this region, the proportion of mutations within a given sample may be small and therefore difficult to detect in the high background of a normal sequence.45 JAK2 exon 12 mutations have been observed primarily in younger patients and in patients with isolated erythrocytosis, and thus may represent a somewhat different phenotype from JAK2V617F-positive PV.
PV is distinguished by the fact that erythroid cells proliferate even in the absence of normal levels of erythropoietin; thus, one would expect that at high hematocrit levels, the production of erythropoietin would be inhibited and serum levels consequently reduced. Indeed, several studies have documented serum erythropoietin levels below the normal reference range in patients with PV.143,144,145
Patients with secondary polycythemia usually have normal to elevated erythropoietin levels, although considerable overlap exists in the range of erythropoietin levels between patients with PV and those with secondary polycythemia rendering the test of marginal value in distinguishing between diagnostic possibilties.144,146 An elevated erythropoietin level generally excludes the diagnosis of PV, but a low erythropoietin level is not pathognomonic of PV; patients with primary familial and congenital polycythemia (PFCP) have levels of erythropoietin that are as low or lower than in PV,147 and instances of normal erythropoietin levels occur in PV patients without apparent explanation. The latter is more likely to occur in PV patients with exon 12 JAK2 mutations.44
In vitro assays of erythroid progenitor cells permit the study of their responsiveness to erythropoietin. In PV patient samples, erythroid BFU-E progenitors grow in culture without added erythropoietin,17 forming colonies that are termed EECs. Detection of EECs in cultures of blood or marrow had previously been the most specific test for PV.12,14,148 In one study, all patients with PV, but none with secondary or other causes of polycythemia, formed EECs in vitro.149
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


