Plasma cell tumors
Noopur Raje, MD  Teru Hideshima, MD, PhD
Teru Hideshima, MD, PhD  Andrew J. Yee, MD
Andrew J. Yee, MD  Kenneth C. Anderson, MD
Kenneth C. Anderson, MD
Overview
Plasma cell disorders have in common a proliferation of monoclonal plasma cells associated with the production of a monoclonal protein. These disorders range from the common, indolent condition of monoclonal gammopathy of undetermined significance to malignancies, such as multiple myeloma, characterized by the presence of hypercalcemia, anemia, renal dysfunction, and/or lytic lesions. Progress in the understanding of the molecular underpinnings of myeloma has led to remarkable advances in its treatment. High-dose melphalan with autologous stem-cell transplant was historically a mainstay of treatment. Now, highly effective and well-tolerated drug classes such as the proteasome inhibitors (e.g., bortezomib and carfilzomib) and immunomodulatory drugs (e.g., lenalidomide and pomalidomide) have rapidly transformed the treatment of myeloma and significantly improved the overall survival. The increasing use of extended treatment strategies such as maintenance therapy and the arrival of newer drug classes such as plasma cell-specific monoclonal antibodies are setting the stage for improving outcomes further.
Multiple myeloma
Multiple myeloma (MM) is a malignant proliferation of plasma cells and plasmacytoid cells in the bone marrow (BM) characterized nearly always by the presence, in the serum and/or urine, of a monoclonal immunoglobulin (Ig) or Ig fragment.1 This disease has probably been recognized since 1845, when the first patient was noted with bone pain and heat soluble “animal matter” in urine.2, 3 The term MM was coined in 1873, reflecting distinct sites of BM involvement. The plasma cell was discovered in 1890 and MM associated with plasmacytosis shortly thereafter, in 1900. The application of electrophoresis in 1939 and immunoelectrophoresis in 1953 allowed for the identification of monotypic Ig characteristic of MM.4, 5
Diagnostic criteria
Active MM is both a clinical and a pathological diagnosis that is defined by the presence of a monoclonal protein in the serum and/or urine, with ≥10% plasma cells in the BM, and associated hypercalcemia, renal dysfunction, anemia, and/or bone disease (also known as the “CRAB” criteria, see Figure 1), related to the MM (Table 1).7, 8 Active MM must be distinguished from other disorders characterized by monoclonal gammopathies, both malignant and otherwise, in particular monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM. Other conditions associated with a monoclonal protein include Waldenström’s macroglobulinemia, non-Hodgkin’s lymphoma, primary amyloidosis, idiopathic cold agglutinin disease, essential cryoglobulinemia, and heavy chain disease. Smoldering MM is defined by the presence of a monoclonal protein ≥3 g/dL and/or ≥10% plasma cells in the BM and the absence of end organ involvement (i.e., the CRAB criteria of hypercalcemia, renal insufficiency, anemia, or bone lesions). The International Myeloma Working Group (IMWG) has recently updated the definition of symptomatic myeloma to include any one of the following biomarkers of disease such as ≥60% marrow infiltration with plasma cells, an involved : uninvolved serum free light-chain (FLC) ratio of ≥100, or >1 focal lesion on an MRI.6
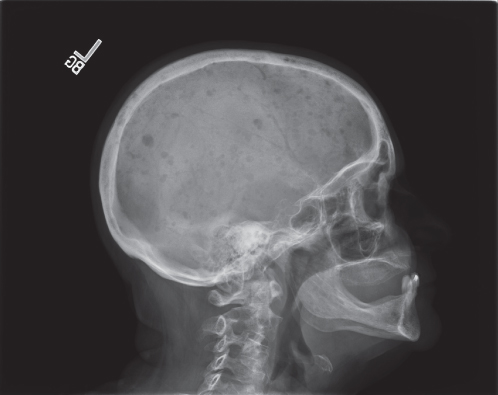
Figure 1 Characteristic lytic bone lesions in multiple myeloma.
Table 1 Classification of monoclonal gammopathies
| Monoclonal gammopathy of unknown significance <3 g/dL M spike and <10% clonal plasma cells in bone marrow and Absence of CRABa criteria Smoldering multiple myeloma ≥3 g/dL Monoclonal protein or 10–60% clonal plasma cells in bone marrow Absence of CRAB criteria Active multiple myeloma ≥10% plasma cells or biopsy proven plasmacytoma and Presence of any of the CRAB criteria or in absence of CRAB criteria: >1 focal lesion on MRI, ≥ 60% plasma cells in bone marrow; or an abnormal involved:uninvolved FLC ratio of ≥100 |
a CRAB criteria is end organ damage due to the plasma-cell disorder: hypercalcemia (>1 mg/dL higher than the upper limit of normal or >11 mg/dL); renal insufficiency (creatinine clearance <40 mL/min or creatinine >2 mg/dL); anemia (hemoglobin >2 g/dL below lower limit of normal or hemoglobin <10 g/dL); or bone lesions (one or more osteolytic lesions on skeletal radiography, CT, or PET CT).
Source: Rajkumar et al. 2014.6 Reproduced with permission of Elsevier.
Monoclonal gammopathy of undetermined significance
MGUS is present in 3.2% of persons 50 years of age or older and 5.3% of persons 70 years of age or older.9 Patients have <3 g/dL monoclonal Ig, fewer than 10% monoclonal marrow plasma cells, and no bone lesions, anemia, hypercalcemia, or renal dysfunction (CRAB criteria).8 In a large experience of 1384 patients diagnosed at the Mayo Clinic, 115 patients progressed to MM, IgM lymphoma, primary amyloidosis, macroglobulinemia, chronic lymphocytic leukemia, or plasmacytoma with relative risk of progression of 25.0, 2.4, 8.4, 46.0, 0.9, and 8.5, respectively.10 The risk of progression of MGUS to MM or related disorders is about 1% per year, and the initial concentration of serum monoclonal protein was a significant predictor of progression at 20 years. Similar results have been published by Cesana et al.11 Independent prognostic factors associated with MGUS transformation to MM include (1) >5% BM plasmacytosis, (2) Bence Jones proteinuria, (3) decrease in polyclonal serum immunoglobulin, and (4) an elevated erythrocyte sedimentation rate.11 More recently, risk factors for progression include an abnormal serum kappa : lambda FLC ratio, a high serum monoclonal protein level ≥1.5 g/dL, and non-IgG type monoclonal protein.12 The risk of progression from smoldering MM to symptomatic disease is related to the proportion of BM plasma cells and serum monoclonal protein level at diagnosis13 as well as abnormal serum FLC ratio.14 In some cases, MGUS can be associated with symptomatology requiring therapy. For example, plasma exchange appears to be efficacious in neuropathy associated with IgG or IgA MGUS.15
Epidemiology
In addition to MGUS, a potential risk factor for the development of MM includes exposure to irradiation or petroleum products. Unlike leukemia, there is no increased risk of MM with benzene exposure.16 Families with two or more affected individuals have been reported, suggesting a possible genetic predisposition.17 MM has also been found to occur with somewhat greater frequency (but less than twofold) in farmers, paper producers, furniture manufacturers, and wood workers.
There are two major misconceptions regarding MM. The first is that MM is a rare disease. MM is the second most common hematologic malignancy, accounting for 24,050 new cancer cases in the United States in the year 2014 and approximately 2% of cancer-related deaths.18 The highest incidence rates have been reported for African-Americans and Pacific Islanders; Europeans and North American white people have intermediate rates, while generally low rates have been reported for Asians living in Asia and the United States.19 Although it has been suggested that the incidence of MM is increasing, data from Olmstead County, Minnesota, demonstrate that the incidence of MM has not changed significantly during the past 46 years.20 A second misunderstanding is that MM is solely a disease of the elderly. In a large Mayo Clinic series, the median age was 66.21, 22 Although 98% of MM patients were 40 years of age or older, 30% of patients were <60 years old. The fact that a significant population of affected individuals is younger than age 70 and therefore can tolerate more aggressive therapeutic approaches influences potential treatment strategies.
Clinical features
The presenting features of 1027 cases of newly diagnosed MM evaluated from 1985 to 1998 are summarized in Table 2.21 Symptoms of bone pain and anemia remain the most common presenting features.
Table 2 Presenting features of multiple myeloma
| Presenting feature | |
| Anemia (hemoglobin ≤12 g/dL) | 73% |
| Calcium ≥ 11 mg/dL | 13% |
| Creatinine ≥ 2 g/dL | 19% |
| Radiographic abnormality (on plain film)a | 79% |
| Bone pain | 58% |
| Fatigue | 32% |
| Weight loss | 24% |
a Lytic lesions present in 67% of patients.
Source: Kyle et al. 2003.21 Reproduced with permission of Elsevier.
Laboratory features
Laboratory evaluation identifies roentgenographic abnormalities in bone and monoclonal Ig in serum and/or urine in the majority of cases. In most series, 50–60% of patients with MM have both serum and urinary monoclonal protein; 20–30% of patients have serum without urinary protein; 15–20% of patients have monoclonal protein in urine only; and only 1–2% of patients do not secrete monoclonal protein in blood and/or urine.23 IgG or IgA monoclonal proteins are most common, and IgD or IgE are rare. A biclonal process is much more common than previously appreciated, often only documented by immunofixation techniques. Thirty-three percent are IgG and IgA; 24% are IgM and IgG. It appears that patients with biclonal and IgD disease have prognoses similar to those patients with monoclonal disease.24 Close observation is an appropriate choice for patients with MGUS or smoldering MM, whereas multiple regimens have been employed for therapy of individuals with overt MM. The natural history of MM is a progressive increase in tumor growth. The M protein doubling time, a measure of the MM growth rate, shortens with each relapse. Eventually, marrow failure develops, with sideroblastic anemia, leukopenia, and thrombocytopenia. The median interval from marrow failure to death is 3 (range 1–9) months.25 Infection and renal failure account for 52% and 21% of deaths, respectively, in patients with MM.26 Acute myeloid leukemia develops in a small fraction of patients but in excess of the anticipated baseline incidence.25
Biology
Cell surface phenotype
B-cell-restricted and associated antigens (Ags) have been utilized to delineate stages of normal and malignant B-cell differentiation.27 Moreover, antigenic profiles are useful not only to identify stages of malignant B-cell differentiation but also to categorize B-cell tumors. MM cells share cell surface expression of some Ags, for example, CD38 and PCA-1 (prostate cancer antigen-1), which are also present on normal plasma cells, suggesting that the normal cellular counterpart of MM is the normal plasma cell. However, a number of other Ags to date have been described on the surface of MM cells, which in some cases react with B cells at stages of differentiation earlier than the plasma cell and also react with non-B cells.28–33 Harada et al.34 have shown that normal plasma cells are CD19+CD56−, whereas no MM cells have this phenotype. The core protein of MUC-1 antigen is expressed on MM cells35 and its inhibition triggers MM cell death.36 The expression and function of adhesion molecules on MM cells is described in the following section. This observed heterogeneity in cell surface phenotype has led to controversy as to the cellular origin of MM.
Cellular origin of MM
As is well known, the cells that accumulate in the BM of patients with MM have plasma cell or plasmablast morphology. However, it has been known since the 1970s, based on studies using anti-idiotypic antibodies, that unique idiotypic determinants can identify clones of peripheral blood lymphocytes in patients with macroglobulinemia, MM, MGUS, and chronic lymphocytic leukemia.37 The presence of idiotypic determinants on cytoplasmic μ-containing pre-B cells in MM BM provided further evidence that the oncogenic event may occur at the pre–B-cell stage. Studies identified B and T cells bearing identical idiotypic determinants, suggesting that target cells for oncogenic transformation could be precursor cells for both B and T cell clones.38 Aneuploid marrow MM cells can express mRNA for cell surface proteins characteristic of myeloid, erythroid, and platelet lineages, also supporting the view that the malignant clone can extend from an early stage of differentiation.39 Moreover, monoclonal B lineage cells in peripheral blood of MM patients, which are late-stage B cells (low CD19 and CD20, moderate CALLA and PCA-1, with strong CD45RO antigen expression) are continuously progressing toward the plasma cell stage.31 However, it remains unclear as to which cell within the malignant clone is “clonogenic” and capable of self-renewal. Some evidence suggests that pre-B and naive B cells migrate from the BM to the lymph node (LN) where antigen recognition, selection, and somatic hypermutation occur. The memory B-cell compartment is thought to contain the cytoplasmic μ-positive precursor cell of MM, which then undergoes Ig class switching in the LN.40 Ig variable (VH) gene sequence analysis has shown MM tumor cells to be postfollicular, with the mutated homogeneous clonal sequences indicating no continuing exposure to somatic hypermutation mechanism.41 VH gene analysis of IgM MM indicates an origin from a memory cell undergoing isotype switch events.42 Mutated heterogeneous sequences in MGUS suggest that tumor cells remain under the influence of the mutator.43 Abnormalities of 14q (the location of IgH) are most common in MM. As proto-oncogenes are translocated to this region and overexpressed in B-cell malignancies including follicular lymphoma, Burkitt’s lymphoma, and chronic lymphocytic leukemia, they may also play a role in the oncogenesis of MM. In addition, translocations involving switch regions indicate that the final oncogenic molecular event in MM occurs late in B-cell ontogeny.43 More recently, CD138− cells with a memory B-cell phenotype are thought to be the clonogenic MM “stem” cells, although this concept needs further validation.44
Role of adhesion molecules, cytokines, and BM stromal cells in MM
Adhesion molecules mediate both homotypic and heterotypic adhesion of tumor cells to either extracellular matrix (ECM) proteins or bone marrow stromal cells (BMSCs) (Figure 2).45 Moreover, they play a critical role in pathogenesis of disease progression. After class switching in the LN, adhesion molecules such as CD44, VLA-4 (very late antigen-4), VLA-5, LFA-1 (leukocyte function-associated antigen-1), CD56, syndecan-1 (CD138), and MPC-1 mediate homing of MM cells to the BM.45–49 Subsequently, binding of MM cells occurs to BMSCs, for example, via VLA-4 to VCAM-1 (vascular cellular adhesion molecule-1), and to ECM, for example, via syndecan to type I collagen and VLA-4 to fibronectin. Such binding not only localizes tumor cells in the BM microenvironment but also stimulates interleukin-6 (IL-6) transcription and secretion from BMSCs with related paracrine growth of MM cells.50–52 Moreover, triggering via CD40 found on tumor cells induces IL-6 transcription and secretion, with related autocrine MM cell growth (Figure 3).53 TNF-α upregulates adhesion molecules on MM cells and BMSCs, thereby increasing binding and cell adhesion-mediated drug resistance (CAM-DR).54 Syndecan-1 is a multifunctional regulator of MM cell growth and survival as well as of bone cell differentiation, and elevated serum syndecan-1 correlates with increased tumor cell burden, decreased metalloproteinase-9 activity, and poor prognosis.55–57 It also mediates decreased osteoclast and increased osteoblast differentiation.55 Adhesion also induces matrix metalloproteinase-1, which favors bone resorption and tumor invasion.58 As the disease progresses, the development of plasma-cell leukemia (PCL) is characterized by decreased expression of certain adhesion molecules (e.g., CD56, VLA-5, MPC-1, and syndecan-1), which in turn facilitates tumor cell mobilization. Furthermore, the acquisition of other adhesion molecules on PCL cells, such as CD11b, CD44, and RHAMM, assists transit through endothelium during egress from the BM. Extramedullary spread of MM cells is facilitated by the reappearance of CD56, VLA-5, MPC-1, and syndecan-1. As adhesion molecules play a central role in the pathogenesis of MM, therapeutic strategies targeting these molecules have been developed and tested in animal models; for example, anti-ICAM-1 antibodies have been shown to inhibit tumor development in severe combined immunodeficient (SCID) mice.59 Moreover, a model of MM in SCID mice bearing human fetal bone grafts (SCID-hu mice) provides for the first time an in vivo model for the evaluation of homing of human MM cells to human BM ECM proteins and BMSCs, the biologic sequelae of binding, as well as testing of novel treatments based on interruption of this process.60, 61 MM cells resistant to melphalan and doxorubicin typically overexpress VLA-4, and adherence to ECM proteins such as fibronectin induces CAM-DR, with upregulation of p27Kip1 in tumor cells.62 As we will discuss in the following paragraph, novel agents, including immunomodulatory drugs (IMiDs), such as thalidomide and lenalidomide, and the proteasome inhibitor bortezomib can target both the tumor cell and its BM microenvironment, thereby overcoming CAM-DR.63–67
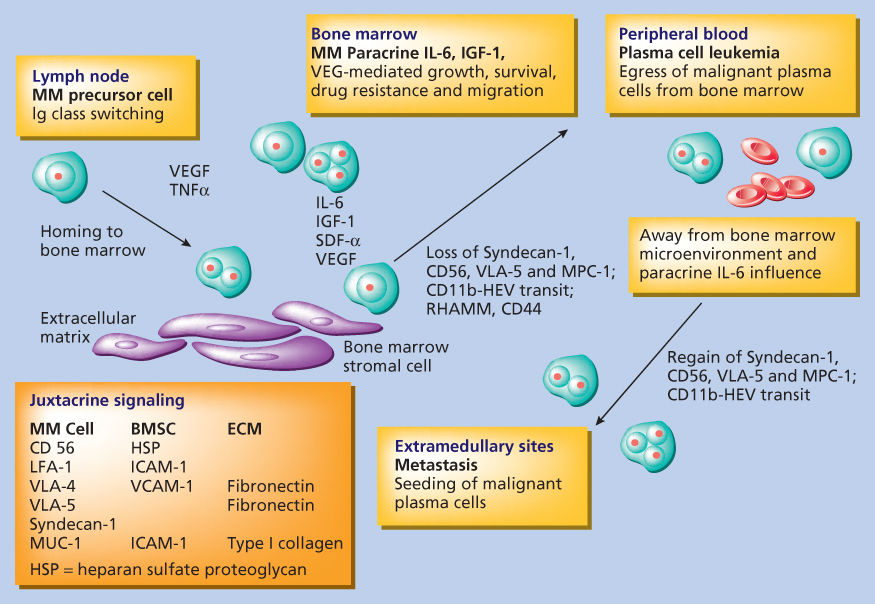
Figure 2 Role of adhesion molecules in myeloma pathogenesis.
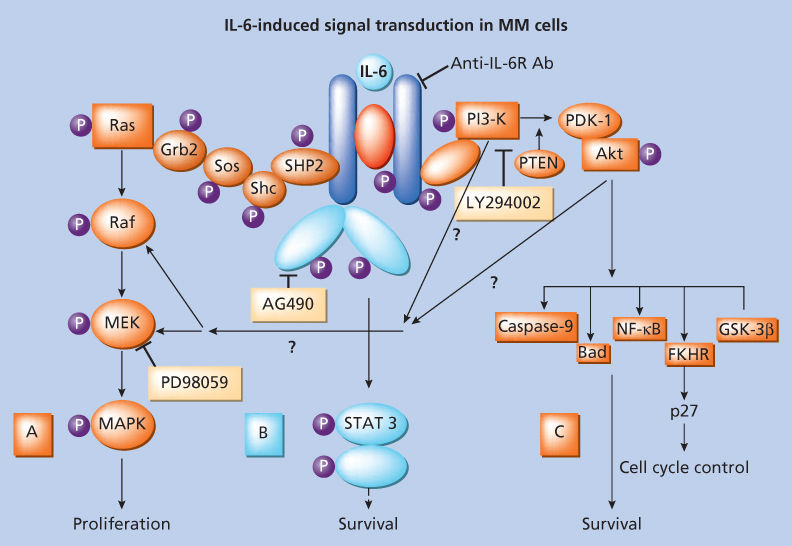
Figure 3 Interleukin-6 signaling cascades.
We have characterized the mechanisms whereby MM cells home to the host BM and adhere to BMSCs and ECM proteins, as well as the functional sequelae of this binding, in order to identify targets for novel therapies. Importantly, our past studies have identified those adhesion molecules mediating MM cell binding to fibronectin and BMSCs, as well as the MM cell growth and survival advantage conferred by this binding.45, 52, 68–70
Our studies show that BMSCs secrete cytokines, such as IL-6,71 insulin-like growth factor-1 (IGF-1),72 vascular endothelial growth factor (VEGF),73, 74 stromal cell derived growth factor (SDF-1α),75 and B-cell activating factor (BAFF),76, 77 which augment MM cell growth, survival, drug resistance, and migration in the BM milieu (Figure 4). Besides localizing tumor cells in the BM microenvironment, our studies demonstrate that adhesion of MM cells to BMSCs also triggers the paracrine NF-κB-dependent transcription and secretion of IL-6 in BMSCs, the major cytokine-mediating MM cell growth, survival, and resistance to dexamethasone-induced apoptosis via activation of p42/44 MAPK, JAK2/STAT3, and PI3K/AKT signaling cascades.50, 52, 72, 78–89 VEGF is secreted by both MM cells and BMSCs, and its secretion is similarly upregulated by binding of MM cells to BMSCs; it augments MM cell growth and BM neovascularization, although the pathophysiologic significance of angiogenesis is undefined.90, 91 VEGF induces migration via PKC signaling.74 Although tumor necrosis factor-α (TNFα) does not directly alter MM cell growth and survival, our studies show that it induces NF-κB-dependent upregulation in cell surface expression of adhesion molecules (ICAM-1 and VCAM-1) on both MM cells and BMSCs, resulting in increased cell adhesion and related induction of IL-6 transcription and secretion in BMSCs.54 Recombinant IL-1β stimulates MM cells to produce IL-6, which consequently augments proliferation of MM cells.92 Transforming growth factor-β (TGF-β) is secreted by MM cells and triggers IL-6 secretion in BMSCs,93 thereby augmenting paracrine IL-6-mediated tumor cell growth. TGF-β secreted by MM cells likely also contributes to the immunodeficiency characteristic of MM by downregulating B cells, T cells, and natural killer cells, without similarly inhibiting the growth of MM cells. IL-10 is a proliferation factor, but not a differentiation factor, for human MM cells.94 IGF-1 has been shown to augment MM cell growth, survival, and drug resistance.72 Macrophage inflammatory protein-1α (MIP-1α) is an osteoclast stimulating factor in MM.95, 96 BAFF is produced by the BMSCs, and specifically by osteoclasts. It signals through several receptors including BAFF-R, transmembrane activator, calcium modulator, and cyclophilin ligand interactor (TACI), and B-cell maturation Ag (BCMA).76, 77 The level of TACI gene expression in MM cells is associated with microenvironment dependence.97 This signaling cascade has a prosurvival effect on MM cells. Autocrine growth mediated by IL-15,98 and most recently IL-21,99 has been demonstrated in both MM cell lines and patient cells.
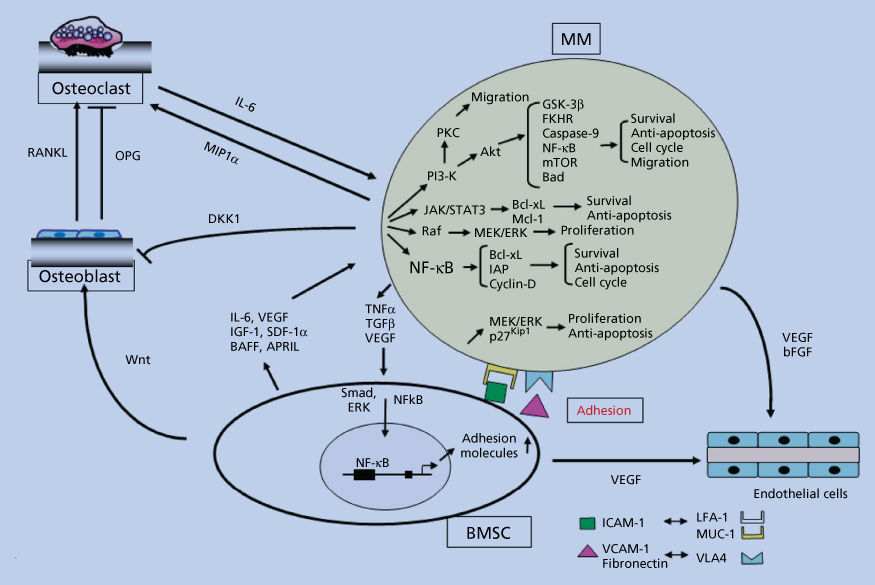
Figure 4 Signaling cascades in the context of the microenvironment.
Wnt signaling regulates various developmental processes and can lead to malignant formation and has been recently studied in the context of MM. Wnts are a family of secreted glycoproteins that bind to frizzled seven-transmembrane span receptors. Intracellularly, the Wnt signaling cascade blocks degradation of β-catenin in proteasomes, thereby leading to accumulation of β-catenin in the cytoplasm. In MM, a canonical Wnt signaling pathway is activated following treatment with Wnt-3a, associated with accumulation of β-catenin. Wnt-3a treatment further led to significant morphological changes in MM cells, accompanied by rearrangement of the actin cytoskeleton.100 Derksen et al.101 demonstrated that MM cells overexpress β-catenin, including its N-terminally unphosphorylated form, consistent with active β-catenin/T-cell factor-mediated transcription. Further accumulation and nuclear localization of β-catenin, and/or increased cell proliferation, was achieved by stimulation of Wnt signaling with Wnt-3a, LiCl, or the constitutively active mutant of β-catenin. Wnt signaling has also been shown as an important regulatory pathway in the osteoblast differentiation of mesenchymal stem cells. Interestingly, MM cells in BM-biopsy specimens contained detectable Dickkopf 1 (DKK1), a negative regulator of Wnt signaling cascade and a target of the β-catenin/TCF pathway.102 Moreover, elevated DKK1 levels in BM plasma and peripheral blood from patients with MM correlated with the DKK1 gene-expression patterns and were associated with the presence of focal bone lesions.103
Most importantly, adhesion of MM cells to BMSCs induces changes in gene profile: that is, upregulation of growth, survival, and drug resistance genes in tumor cells; upregulation of adhesions molecules on MM cells and BMSCs; and changes in cytokines in BMSCs both in in vitro and in vivo models of human MM in mice.104–106 Interaction of MM cells with BMSCs activates Notch signaling, which induces melphalan resistance.107 Induction of proteasome activity when MM cells bind to BMSCs may sensitize them to therapy.
We have shown that IMiDs (e.g., lenalidomide) inhibit VEGF and IL-6, which are known to downregulate antigen-presenting function of dendritic cells in MM.108 Moreover, lenalidomide directly activates CD28 on T cells, thereby stimulating transcription and secretion of IL-2, with resultant upregulation of T and NK cell anti-MM activity.108, 109 Lenalidomide can upregulate antibody-dependent cellular cytotoxicity (ADCC).110 IMiDs also affect the cytokine signaling stimulated by the interaction of effector cells with MM cells and BMSCs, and this is via regulation of SOCS1, a member of the suppressor of cytokine signaling (SOCS) genes.111
Molecular pathogenesis of MM
The malignant plasma cells in MM are localized to the BM in close association with BMSCs. They are long-lived cells with a very low (1–2%) labeling index (LI) that provides a measure of the proliferative rate of the malignant BMPC predicting survival in patients with newly diagnosed MM. The rearranged Ig genes are extensively somatically hypermutated in a manner compatible with antigen selection, with no evidence that the process of hypermutation is continuing.41 However, MM cells have a significantly lower rate of Ig secretion than normal plasma cells. Thus, it appears that the critical oncogenic events in MM cells either occur after or do not interfere with most of the normal differentiation process involved in generating a long-lived plasma cell.
Gene expression profiling has recently been utilized to characterize changes associated with the progression from normal plasma cells to MGUS to MM.112–114 The mRNA profile of MGUS and MM is similar and distinct from that of normal plasma cells.114 These studies may not only enhance understanding of basic pathophysiology but also identify novel therapeutic targets.
By conventional analyses, karyotypic abnormalities are detected in MM at a frequency of 30–50% in large studies of MM tumors (Table 3).115–117 The frequency and extent of karyotypic abnormalities correlates with the stage, prognosis, and response to therapy. For example, approximately 20% are abnormal in stage I disease, 60% in stage III patients, and >80% for extramedullary tumor. This analysis is dependent on obtaining reliable metaphase preparations and greatly underrepresents the extent of DNA alterations in these infrequently dividing cell populations. By interphase fluorescence in situ hybridization (FISH) analysis, two studies report that at least one chromosome is trisomic in 96% or 89% of MM tumor samples, respectively.118, 119 Although conventional karyotypes are not routinely reported for MGUS, it appears that a substantial fraction of MGUS plasma cells are aneuploid as well. By FISH analysis, the incidence of trisomy for at least one chromosome was 43% and 53% in two studies of MGUS cells; in the former, 61% of the cells had an aneuploid DNA content by image analysis.118, 120 The characteristic numerical abnormalities are monosomy 13 and trisomies of chromosomes 3, 5, 7, 9, 11, 15, and 19. Nonrandom structural abnormalities most frequently involve chromosome 1 with no apparent locus specificity; 14q32(IgH) locus occurs in 20–40%; 11q13(bcl-1 locus) in about 20%, but mostly translocated to 14q32; 13q14 interstitial deletion in 15%; and 8q24 in about 10%, with about half of these involved in a translocation. Importantly, a recent report documents similar translocations in MGUS and MM, including t(4;14)(p16.3;q32) and t(14;16)(q32;q23), without any obvious clinical or biologic correlation.121
Table 3 Myeloma chromosomal alterations
| Genetic lesion | Incidence (%) |
| Hyperdiploid | 60 |
| t(11;14) | 20 |
| t(4;14) | 15 |
| MAF translocations | 5 |
| Del (13q)/monosomy 13 | 50–60 |
| Del (1p) | 7–40 |
| Chr 1q21 amplification | 40 |
| Cyclin D dysregulation | 80 |
| RAS mutations | 30–50 |
| FAM46C, DIS3 | 10–21 |
| NF-κB activating mutations and CNVs | 15–20 |
| IgH MYC rearrangements | 15 |
| UTX deletions and mutations | 30 |
| TP53 inactivations (mutations+del(17p)) | 10–20 |
| p18 and/or Rb inactivation | <5 |
| p14 promoter methylation | <5 |
| PTEN loss | <2 |
The hallmark genetic lesion in many B-lymphocyte tumors involves dysregulation of an oncogene as a consequence of a translocation involving the IgH locus (14q32.3); less frequently, variant translocations involve one of the IgL loci (2p12, kappa or 22q11, lambda) (Tables 3 and 4). From conventional karyotypic analyses, translocations involving 14q32 appear to occur in about 20–40% of MM with an abnormal karyotype.123 The incidence of these translocations is significantly higher in the extramedullary phase of the disease and in cell lines, perhaps because of a higher number of metaphase spreads that are examined. In about 30% of these translocations, the partner chromosomal locus is 11q13 (bcl-1, cyclin D1), but in most cases, the partner is not identified (14q32+). Transcriptional activation of cyclin D1 has recently been confirmed in some primary tumors, as has cyclin D3 activation associated with t(6;14)(p21;q32) translocation.121, 124, 125 Other recurrent partner loci have been identified infrequently, including 8q24(c-myc) in <5%, 18q21(bcl-2), 11q23(MLL-1), and 6p21.1. By combining conventional karyotypic analysis with a comprehensive Southern blot assay, which detects translocations involving IgH switch regions, it has become apparent that most MM cell lines and one primary tumor fully examined have IgH translocations that mainly involve IgH switch regions.43, 126 Recent FISH studies have also shown that IGH gene rearrangements are present in 73% of MM patients.127 The apparent oncogene dysregulated by t(4;14) is the fibroblast growth factor receptor 3 (FGFR3) gene, and it is possible that dysregulated expression of FGFR3, as a result of t(4;14), receives an FGFR3-mediated signal from FGF produced by stromal cells in the BM microenvironment.128 In addition to FGFR3, the t(4;14) translocation in MM regulates a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts.129 Ectopic expression of FGFR3 promotes MM cell proliferation and prevents apoptosis, and its oncogenic potential has been tested in a murine model, confirming its capacity to transform hematopoietic cells.130, 131 Finally, there is evidence that elevated expression of c-myc and selective expression of one c-myc allele may occur frequently in MM, even though structural genetic changes near c-myc have been identified in only 10–20% of tumors.
Table 4 Nonimmunoglobulin sites for illegitimate switch recombination
| Chromosome | Gene | Function |
| 11q13 | Cyclin D1 | Induces growth |
| 4p16 | FGFR3, MMSET | Growth factor |
| 8q24 | MYC | Growth/apoptosis |
| 16q23 | c-maf | Transcription factor |
| 6p25 | IRF4 | Transcription factor |
Source: Kuel and Bergsagel 2002.122 Reproduced with permission of Nature Publishing Group.
Ras mutations occur in about 39% of newly diagnosed MM patients, and the frequency of ras mutations increase with disease progression. Mutations of N- and K-ras are rarely detected in solitary plasmacytoma and MGUS, but occur more frequently in MM (9–30%) and in the majority of terminal disease or PCL patients (63–70%).132, 133 Activating mutations of the ras oncogenes may also result in growth factor independence and suppression of apoptosis in MM.
Although translocation (14;18) occurs at a low frequency (0–15%) in MM, an overexpression of Bcl-2 is seen in the majority of MM patients and in MM cell lines.134, 135 High levels of Bcl-2 protein are likely to mediate the resistance of MM cells to apoptosis induced by IL-6 deprivation, staurosporine, or other drugs.136 In a murine MM cell line, Bcl-XL showed a predominant role in preventing apoptosis in response to cycloheximide treatment or IL-6 withdrawal.137 Similarly, overexpression of Bcl-2 or Bcl-XL could prevent apoptosis induced by IL-6 withdrawal in the B-9 IL-6-dependent cell line.138 Mcl-1 is overexpressed in MM cells, upregulated by IL-6, and mediates potent resistance to apoptosis, whereas Mcl-1 downregulation triggers apoptosis (Figure 5).140
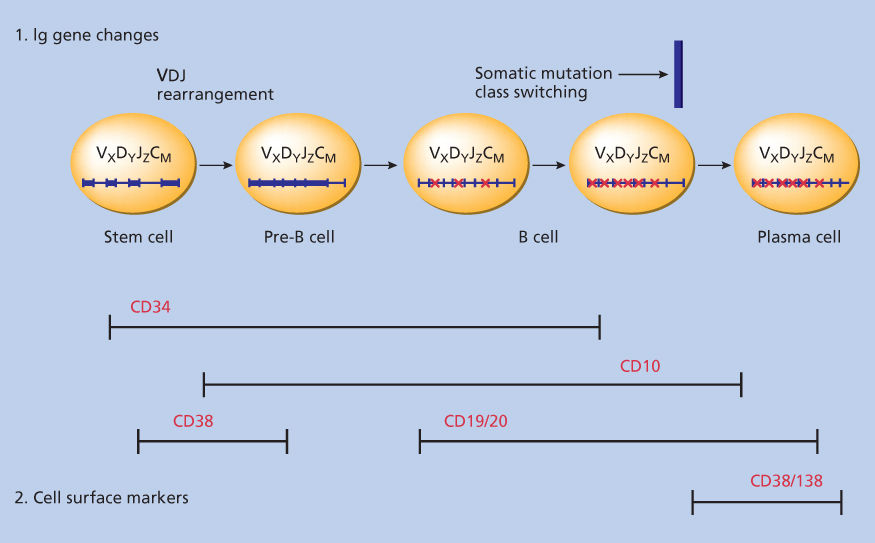
Figure 5 Apoptotic signaling pathways.
Source: Adapted from Hideshima et al. 2014.139
Chromosome 13 deletions are present in over 50% of MM and are associated with poor prognosis.121, 141–143 However, these deletions are also associated with MGUS, and their role in transformation to MM is therefore at present undefined.121, 144, 145
Recent understanding of the molecular pathogenesis of MM has resulted in a new proposed classification of MM.146, 147 Majority of MM tumors have chromosomal abnormalities and are broadly classified into hyperdiploid (HRD) or nonhyperdiploid (NHRD) tumors. Nearly half of MM tumors are HRD, the remaining being categorized as NHRD, which includes tumors that are hypodiploid, pseudodiploid, or subtetraploid. These NHRD tumors have been associated with a poorer prognosis. Five recurrent IgH translocations have been seen in MM including MMSET and FGFR3 (15%), cyclin D3 (3%), cyclin D1 (15%), c-maf (5%), and MAFB (2%) accounting for a prevalence of 40%. Recent evidence suggests that three of these five translocations are predominant in the NHRD tumors.
Deletions of chromosome 17p involving the TP53 locus are rare in newly diagnosed myeloma (5–10%), but more common in relapsed and refractory cases (20–40%), and are associated with a negative prognosis.148, 149 Regimens containing bortezomib can however overcome this unfavorable prognosis, as shown by the HOVON-65/GMMG-HD4 trial.150 1q21 amplification is detected by FISH in about 40% of newly diagnosed and 70% of relapsed myeloma. It can negatively impact overall survival (OS). Possible downstream target genes include CKS1B, a protein that regulates cyclin-dependent protein kinases, PSMD4, a proteasome subunit modulating response to bortezomib treatment, MCL1 and BCL9.151–153 In addition, deletions of 1p, present in 7–40% of patients, are linked to reduced PFS (progression-free survival) and OS despite autologous stem-cell transplant.154–156 FAM46C loss or FAM46C mutations (evident in 15% of patients) are especially associated with shortened survival (median OS 25.7 months vs 51.3 months, P = 0.004).156, 157 The biologic function of FAM46C is unknown, but possibly related to mRNA stabilization. Other abnormalities include MYC rearrangements involving unbalanced translocations and insertions, small duplications, amplifications, and inversions on chromosome 8p24158–161; homozygous deletions of 11q22 locus resulting in loss of YAP1, BIRC3, and BIRC2 genomic region162–164; chromosome 4, 14, and 16 aberrations, disrupting FGFR3, WWOX, and CYLD; and deletions or amplifications of chromosome 6 and homozygous deletions of Xp11.2 locus,165, 166 involving UTX, a histone H3 lysine 27 (H3K27) demethylase mutated in 10% myeloma.157, 167
With the advent of cDNA microarrays, an expeditious and comprehensive gene expression profiling is possible to better define disease biology, highlight prognostic factors, and identify potential targets for novel therapies.124, 168 These studies may also identify mechanisms of sensitivity versus resistance to conventional and novel MM therapies.169, 170 Known target antigens likely represent only the tip of the iceberg. Most recently, array comparative genomic hybridization (aCGH) has been correlated with gene profiling to identify chromosomal amplifications and transcript overexpression, respectively.165 Classical overexpression and knockdown experiments may be done first in cancer models and then in MM models to identify potential novel targets for monoclonal antibody (cell surface) or small molecule inhibitor (intracellular) therapies.
Somatic mutations and interclonal diversity
MM is often preceded by MGUS or smoldering myeloma transforming to overt myeloma,171, 172 characterized by accumulation of mutations conferring growth advantage (driver mutations) or functionally irrelevant mutations (passenger mutations). To date, more than 300 myeloma patient DNA samples have been sequenced using whole-genome-sequencing or whole-exome sequencing approaches.157, 173, 174 Frequently mutated genes include KRAS, NRAS, FAM46C, DIS3, and TP53. Other significant genes are BRAF (4% of patients), TRAF3, CYLD, RB1, PRDM1, and ACTG1. TRAF3 and CYLD mutations, together with homozygous deletions in BIRC2/BIRC3, NIK overexpression, and mutations in other genes (CARD11 and MYD88) contribute to constitutively activating the NF-κB pathway. Genes involved in protein homeostasis, unfolded protein response, or lymphoid/plasma-cell development, such as PRDM1 involved in plasmacytic differentiation, and XBP1, IRF4, LRRK2, SP140, and LTB form a cluster of genes mutated in myeloma. Other recurrent mutated genes are ROBO1, a transmembrane receptor involved in β-catenin and MET signaling, EGR1 transcription factor, FAT3, a transmembrane protein belonging to cadherin superfamily, and histone-modifying genes (MLL, MLL2, MLL3, WHSC1/MMSET, WHSC1L1, and UTX among others). -PCL patients tend to have different types of aberrancies, such as p14ARF promoter methylation, PTEN loss, RB1 mutations, and higher rates of TP53 mutations and deletions.175
Recent data supports the concept of intratumor heterogeneity in myeloma, where different subclones can emerge and become predominant following different mechanisms of evolution, including linear, branching, parallel, or convergent evolution.173, 176 Clonal diversity similar to Darwinian-like selection favors cancer progression and adaptation to therapy. Next-generation sequencing analyses show that most patients have a subclonal structure at diagnosis, with one predominant clone and several others which can reappear at different stages of disease evolution or following treatment.173, 177, 178
Prognostic factors
Multiple attempts have been made to define clinical and laboratory parameters that have prognostic significance.7, 179–181 The Durie–Salmon system was historically utilized (Table 5).7 Tumor cell mass for patients in stage I is low at <0.6 × 1012 cells/m2, intermediate for patients with stage II disease at 0.6 to 1.2 × 1012 cells/m2, and high for patients with stage III disease with >1.2 × 1012 cells/m2. In this system, survival duration is 61.2, 54.5, 30.1, and 14.7 months for patients with stage IA, stage IB + IIA + IIB, stage IIIA, and stage IIIB disease, respectively.
Table 5 Durie–Salmon staging system and international staging system
| Stage | Durie–Salmon stage | ISS stage | Overall survival by ISS stage |
| I | All of the following:
| Serum β2 microglobulin <3.5 mg/L and Serum albumin ≥3.5 g/dL | 62 months |
| II | Neither stage I nor stage III | Serum β2-microglobulin <3.5 mg/L, but serum albumin <3.5 g/dL or Serum β2-microglobulin 3.5 to <5.5 mg/L, irrespective of serum albumin | 44 months |
| III | One or more of the following:
| Serum β2-microglobulin ≥5.5 mg/L | 29 months |
Source: Durie and Salmon 19757 and Greipp et al.181
Many additional single parameters have been examined for their value as prognostic features. Higher labeling indices, serum IL-6 receptor levels, more ras mutations, more aggressive disease, and shortened survival have been reported in patients with plasmablast morphology.182 Serum β2 microglobulin (β2M) represents the light chain of the major histocompatibility complex of the cell membrane, and increased serum β2M results from release by tumors with high growth fraction and cell turnover rates. In patients with MM and normal renal function, rising serum β2M predicts for progression.183 The LI, a measure of DNA synthesis by MM cells, predicts for survival. It is usually low (<1%) at diagnosis, higher at relapse, and lower in MGUS and indolent MM.184 Chromosome 13 deletions are present in over 50% of MM and are associated with poor prognosis141–143; however, these deletions are also associated with MGUS,121, 144, 145 and their role in transformation to MM is therefore at present undefined. HRD MM has improved outcome and distinct clinical features, and chromosome 13 deletion does not have adverse impact.185 Moreover, it is not prognostic for response to bortezomib, highlighting the importance of prognostic factors for particular therapies.186 Gene expression profiling will not only define disease pathogenesis but also identify both novel prognostic factors and potential therapeutic targets.124, 130 These studies will also identify mechanisms of sensitivity versus resistance to conventional and novel MM therapies.169, 170 Cyclin D dysregulation has been identified as an early and unifying event in MM. Using gene expression profiling to identify five recurrent translocations, specific trisomies, and expression of cyclin D2 genes, MM can prognostically be divided into eight TC (translocation/cyclin D) groups.187 Additional molecular classifications have been proposed,188 with high-risk myeloma defined by deregulated expression of genes mapping to chromosome 1.189 Most recently, the first DNA-based classification scheme has been proposed to predict outcome to high-dose therapy (HDT).165
Serum IL-6 levels in some studies appear to correlate both with stage of disease and survival.190, 191 IL-6 stimulates hepatocytes to produce acute phase proteins, such as CRP; CRP therefore may reflect the IL-6 level and proliferative status of BM plasma cells. Indeed, CRP levels are significantly lower in patients with MGUS than in those with MM, and survival can be correlated with serum CRP level.192 High serum soluble interleukin-6 receptor (sIL-6R),193 hepatocyte growth factor,194 and syndecan-1195 levels, as well as low serum hyaluronate levels,196 are independent prognostic factors predicting poor outcome. The percentage of circulating plasma cells in peripheral blood and their labeling indices are independent prognostic factors for survival in MM after both conventional and HDT.197, 198 Circulating endothelial cells also correlate with disease course and response to thalidomide.199 Finally, circulating proteasome levels are an independent prognostic factor for survival.200
Many of these factors are interrelated and, therefore, of limited independent value. Using multivariate analysis, several groups have found that the best combination of variables to predict outcome was serum β2M, reflecting both the tumor burden and the renal function; and the proliferative activity of plasma cells, evaluated by the LI or number of tumor cells in S-phase. Age and performance status also improves the prognostic assessment.201, 202 An international staging system (ISS) based on serum β2M and albumin has provided a three-stage ISS (Table 5), which is currently the most commonly used staging system and in reporting for clinical trials.181
Complications
Complications of MM include bone disease and hypercalcemia, hyperviscosity, recurrent infections, renal failure, and cardiac dysfunction.
Bone disease and hypercalcemia
The presence of osteolytic bone lesions, bone pain, increased risk of pathological fractures, or generalized bone loss (or osteoporosis) is a well-defined feature of myeloma.203 Myeloma bone disease is characterized by an imbalance between osteoblast and osteoclast activities, with suppression of bone formation by osteoblasts and uncoupled activation of osteoclasts (Figure 6).204, 205 The ligand for receptor activator of NFκB (RANKL) binds to RANK receptor to stimulate osteoclast differentiation, formation, and survival206; myeloma cells produce RANKL and upregulate RANKL expression in BMSCs and osteoblasts via direct contact, signal induction,207–209 or production of IL-7. Moreover, they promote suppression of osteoprotegerin (OPG),210–212 a decoy receptor that normally prevents RANK–RANKL interaction213 via soluble factors, integrin α4β1-VCAM1 interaction,214 production of DKK1,215or inactivation by syndecan-mediated internalization into myeloma cells.216 Interestingly, OPG levels are decreased in the serum of myeloma patients and correlate with the presence of lytic bone lesions217; a high RANKL/OPG ratio is associated with a poor prognosis.218 Recombinant OPG constructs, soluble RANK, OPG peptidomimetics,211, 213, 219 and, more recently, an anti-RANKL antibody, denosumab,220 have been developed to modulate RANKL/OPG axis and reduce osteoclast activity in myeloma. MIP-1α, or chemokine C–C motif ligand, is also produced by myeloma cells and promotes maturation of precursor cells into osteoclasts; MIP-1α signals via CCR1 and CCR5 on osteoclasts and can further upregulate RANKL in stromal cells.96, 221, 222 MIP-1α levels are elevated in myeloma patients,223 while MIP-1α silencing or blockade of CCR1 reduces bone disease in in vitro or animal models.96 IL-6,224 PTHrP,225, 226 annexin II,227 and ephrinB2/EphB4 axis228 also promote bone resorption. Osteoblast suppression is another major player in myeloma bone disease: WNT signaling antagonists, including DKK1, frizzled related protein-2 (FRP-2),229 and sclerostin (SOST),230 interfere with osteoblast maturation. DKK1 is expressed by myeloma cells and can upregulate RANKL levels in osteoblasts, increasing osteoclast activity.103, 231 DKK1 levels are increased in the serum of myeloma patients,232 and anti-DKK1 antibodies have been tested in animal studies233–235 and are currently being studied in clinical trials. Finally, high levels of activin A, a member of TGF-β superfamily, IL-3, and IL-7 via RUNX2/CBFA1 blockade can inhibit bone formation and promote bone reabsorption. Furthermore, the bone niche itself supports myeloma cell survival and prevents TNF-α-mediated apoptosis.236
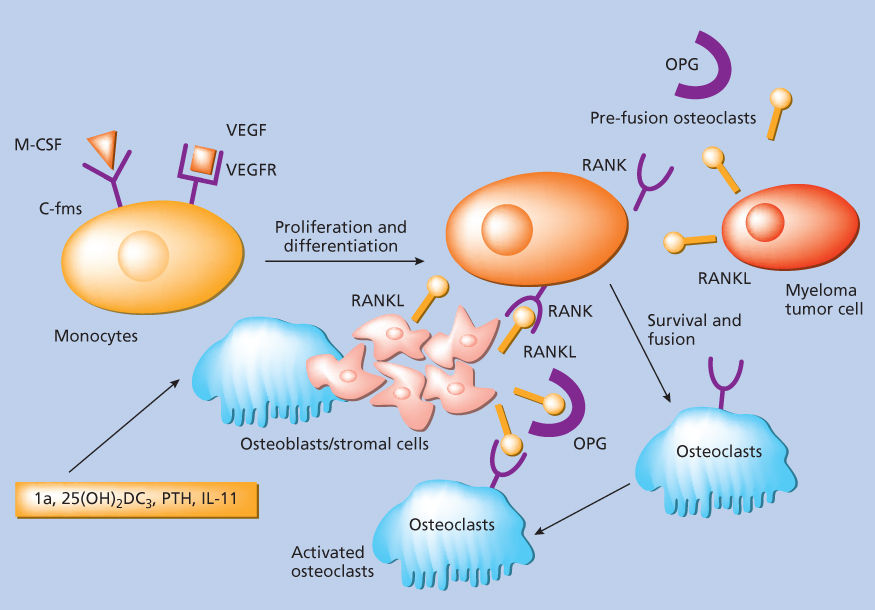
Figure 6 Osteoclastogenesis.
Treatment
Bisphosphonates play a key role in the supportive management of MM by not only blocking osteoclasts and modulate osteoblasts but also have an effect on tumor burden237; a similar response is reported with OPG peptidomimetics and RANKL constructs in in vivo xenograft models.238 Bisphosphonates, especially zoledronic acid, are currently used in the clinic to reduce bone disease,239, 240 but are also associated with an increase in OS when compared to placebo based on a recent meta-analysis.241 Markers of bone resorption and formation correlate with the extent of osteolytic disease.242 Specifically, urine levels of pyridinoline (PYD) and deoxypyridinoline (DPD) cross-links and serum levels of tartrate-resistant acid phosphatase isoform 5b (TRACP-5b), a resorption marker only produced by activated osteoclasts and of collagen degradation products, including the N-terminal cross-linking telopeptide of type I collagen (NTX) are elevated in myeloma patients compared with healthy controls and can predict early progression of bone disease in myeloma. Conversely, bone formation markers, such as bone alkaline phosphatase (bALP) and osteocalcin (OC), are reduced.242 Another randomized trial demonstrated that oral clodronate slowed progressive skeletal disease and associated morbidity but achieved no benefit in survival. Pamidronate has been shown in a prospective randomized trial to reduce skeletal-related events, including pathologic fractures, radiation therapy to bone, and spinal cord compression in patients with Durie–Salmon stage III MM and ≥1 lytic bone lesion.243 This benefit was maintained until 21 months and formed the basis of previous ASCO recommendations, suggesting that MM patients remain on intravenous bisphosphonates indefinitely to reduce skeletal events and pain, regardless of their response to chemotherapy.244, 245 Interestingly, patients in this study who had failed first-line chemotherapy also had improved survival, suggesting that bisphosphonates may have anti-MM activity.246 Recent evidence supports the view that bisphosphonates may downregulate IL-6 production from BMSCs as well as induce apoptosis of both osteoclasts and tumor cells. Pamidronate has been studied in patients with indolent MM. Bone turnover was reduced in treated patients,247, 248 but no significant antitumor activity was noted.249 Zoledronate is more potent than pamidronate and has the advantage of a shorter infusion time with similar efficacy compared to pamidronate for preventing skeletal related events.250 More recently, the MRC Myeloma IX trial has demonstrated the superiority of zoledronic acid compared with the oral bisphosphonate clodronate in patients with MM with an OS benefit favoring zoledronic acid.251 A word of caution should however be exercised with the use of bisphosphonate therapy because of reported cases of osteonecrosis of the jaw.252–254 This newly recognized complication in a small percentage of patients led ASCO to update their guidelines and now recommend discontinuing bisphosphonates after 2 years in patients with very good partial response (VGPR) or better and continuing if response is partial response (PR) or less.255 Other supportive care measures such as vertebroplasty or kyphoplasty can restore supine stability and relieve pain for patients with localized disease.256, 257
The treatment of hypercalcemia consists of treatment of the underlying MM, as well as inhibition of osteoclastic bone resorption with corticosteroids, calcitonin, and/or bisphosphonates. Corticosteroids may impair formation of new osteoclasts. Calcitonin and corticosteroids are almost always effective for the short term and useful even in the setting of renal failure. Bisphosphonates bind to the bone surface and inhibit osteoclast activity, and they constitute a mainstay of treatment. Therapeutic recommendations therefore include initial cytotoxic therapy for MM, saline hydration, and bisphosphonate therapy, with the use of calcitonin for nonresponders to these first-line treatments. In a randomized trial of 287 patients with hypercalcemia of malignancy, zoledronate was found to be superior to pamidronate.258 Hypercalcemia may also be compounded by patient immobility because of bone pain or other reasons.
Hyperviscosity
Hyperviscosity is characterized clinically by spontaneous bleeding with neurologic and ocular disorders. Hyperviscosity occurred in 4.2% of 238 patients with IgG MM and in 22% of 46 patients with serum IgG M components >5.0 g/dL.259 The IgG3 subclass produces hyperviscosity at lower levels than other IgG paraproteins.260 The severity of the syndrome is not directly related to the serum viscosity. Clinical findings improve with plasmapheresis, which reduces both MM protein concentration and serum viscosity.
Recurrent infections
Patients with MM had 15 times more infections than a control group of patients with heart disease.261 Streptococcus pneumoniae and Haemophilus infections usually occur early and typically during response to chemotherapy. Gram-negative infections occur in refractory, advancing disease; in the setting of previous antibiotic therapy; instrumentation; immobilization; colonization with hospital flora; and azotemia. Fatal infections may be hospital-acquired, emphasizing the need to minimize indwelling foreign bodies such as catheters in patients with MM. There is lack of correlation between bacteremia (either gram-negative or -positive) and chemotherapy-induced febrile neutropenia. Fungal, herpes, mycobacterial, and Pneumocystis infections are only rarely described in MM patients. Infection is the most common cause of death (20–50% of cases).26
The evidence of increased clinical infections in MM has led to attempts at prophylaxis. Although MM patients have normal-fold rises in antibody titers after pneumococcal vaccination, preimmunization titers are markedly diminished.262 Postimmunization titers are therefore low and considered nonprotective. Nonetheless, because of its low cost and possible benefit to some patients, the use of pneumococcal vaccination has been recommended. In a double-blind randomized trial of gammaglobulin prophylaxis in patients with MM, no benefit of gammaglobulin prophylaxis at reducing infection was noted,263 although a trial in patients in the plateau phase suggested possible benefit.264 At present, gammaglobulin is reserved for those patients with recurrent or life-threatening infections and hypogammglobulinemia.
Renal failure
Renal failure in MM can predict for adverse outcome. One series found that 22% of patients had a serum creatinine ≥2 mg/dL at diagnosis; renal function normalized with treatment in 48% of those with creatinine <4 mg/dL.265 The causes of renal failure in MM are often multifactorial and include hypercalcemia; MM kidney, with distal and proximal tubules obstructed by large, laminated casts containing albumin, IgG, and κ and λ light chains surrounded by giant cells; hyperuricemia; toxicity from intravenous contrast; dehydration; plasma-cell infiltration; pyelonephritis; and amyloidosis. The most important predisposing factor is dehydration; aggressive hydration is therefore crucial to avoid irreversible renal dysfunction. Otherwise, treatment is for the underlying disease, along with avoidance of intravenous contrast. The type and quantity of proteinuria can distinguish MM kidney, with larger amounts of light chains and less albuminuria; light-chain deposition disease, characterized by low levels of both light chains and albumin in urine; and amyloidosis, in which large amounts of albuminuria and less light-chain proteinuria occur.266
The renal manifestations associated with the production of monoclonal light chains in MM, light-chain deposition disease, and amyloidosis result from the deposition of certain Bence Jones proteins (BJP) as tubular casts, basement-membrane precipitates, or fibrils, respectively.267–269 For unknown reasons, the severity of the renal manifestations varies greatly from patient to patient. BJP from 40 patients were injected into mice and 26 (65%) were deposited in mouse kidneys as tubular casts, basement-membrane precipitates, or crystals in a pattern similar to those noted in patients.269 This experimental model has potential value for the identification and differentiation of nephrotoxic or amyloidogenic light chains. The development of progressive kidney damage and MM kidney has been demonstrated in IL-6 transgenic mice, shedding additional insight into the role of IL-6 in the pathogenesis of MM.270
Cardiac failure
The mean and median age of patients with MM is approximately 60 years, and affected patients are also frequently at risk of cardiovascular disease. However, patients can be uniquely susceptible to cardiac ischemia and/or congestive heart failure (CHF) because of myocardial infiltration with amyloid, causing dilated or restricted cardiomyopathy, hyperviscosity syndrome, and/or anemia. Rarely, MM patients are also susceptible to high output CHF,271 mainly in patients with extensive bone disease.272 This has been attributed to arteriovenous shunting in bone lesions.273
Anemia
Anemia in MM can be due to a number of factors, including tumor infiltration of the BM, renal impairment, the myelosuppressive effects of chemotherapy, and a deficient production of erythropoietin (EPO) relative to the degree of anemia. Pilot studies demonstrated efficacy of exogenous EPO administration in MM.274–276 Osterborg et al.277 have carried out a randomized study of EPO therapy at 10,000 U/d, a titrated dose of EPO starting with 2000 U/d and escalating stepwise until response, or no EPO for 24 weeks; response was defined as an increase in Hb > 2g/dL and elimination of transfusion need. Sixty percent of EPO-treated groups responded, 72% of those with low EPO levels, and only 20% of those with normal EPO levels. 10,000 units SC three times weekly was the optimal starting dosage, although more recently 40,000 units SC once weekly is a commonly used approach. However, erythropoiesis-stimulating agents such as EPO and darbepoietin are restricted under a risk evaluation and mitigation strategy (REMS) program owing to several trials showing increase in thrombotic risk and adverse effects on survival with these drugs (though this risk has not been as well demonstrated in MM compared to solid tumors).278, 279
Neuropathies
A variety of malignant and paraproteinemic disorders can be associated with neuropathies.280 In MM, a symmetric, distal sensory, or sensorimotor neuropathy is most common and is associated with axonal degeneration, with or without amyloid deposition; there is no specific therapy. In some cases, this is associated with monoclonal antibodies directed against peripheral nerve myelin.281
Associated diseases
MM has been described in association with both hematologic disorders and solid tumors. Acute leukemia either is induced by leukemogens, such as radiation and alkylating agents, or is part of the natural history of MM. The mean interval from diagnosis of MM to occurrence of acute leukemia is 60 (17–147) months, consistent with either possibility. The occurrence of acute leukemia in untreated MM patients suggests that it may be part of the natural history of the disease.282 Furthermore, acute leukemia has been reported in 6 of 125 (4.8%) MM patients treated with alkylating agents, which is significantly higher than the incidence of acute leukemia in ovarian cancer patients treated with irradiation and alkylators.25 Actuarial risk of leukemia in MM patients treated with melphalan and prednisone (MP) or with melphalan, cyclophosphamide, carmustine, and prednisone has been reported to be as high as 17.4% at 50 months from initiation of therapy.283 Gonzalez et al.282 described 11 of 476 patients with MM who developed myeloid leukemia or sideroblastic anemia. All had received melphalan–prednisone for a median of 3 years and had major cytogenetic abnormalities. This study suggests that leukemia is predominantly treatment related. Finally, in 628 patients with MM, the incidence and diversity of solid tumors were similar to those observed in otherwise healthy persons of the same age.284
Treatment
While MM is not considered curable, its treatments are very effective and very well tolerated, including in the older patient population. The adoption of high-dose melphalan with autologous stem-cell transplant and the introduction of two new drug classes—IMiDs such as lenalidomide and proteasome inhibitors such as bortezomib—have resulted in dramatic improvements in outcomes. A retrospective study comparing patients diagnosed in 1997 and later to those diagnosed before 1997 found a significant improvement in OS, 44.8 months versus 29.9 months, respectively.285
An area of active investigation is when to initiate treatment for patients with MM. There is uniform consensus recommending treatment for active MM as defined by the presence of one of the CRAB criteria of end organ involvement.6 For patients who are asymptomatic, that is, with smoldering MM, the current paradigm recommends close observation. However, the availability of well-tolerated, effective myeloma therapy has now motivated studies examining treatment earlier in the disease, before the onset of symptoms. Recently, the Spanish myeloma group conducted a randomized study of active treatment with lenalidomide and dexamethasone v. observation in patients with high-risk smoldering MM.286 High risk was defined by the presence of both BM involvement and elevated serum monoclonal protein or flow cytometry criteria and suppressed immunoglobulins (immunoparesis). The study found that 3-year OS was superior in the group undergoing active treatment, 94% versus 80%, and this was the first time this type of benefit was seen in a smoldering MM trial. These results suggest that earlier initiation of therapy may be helpful, and the generalizability of these findings awaits confirmation in further trials as well as identifying the specific patients who are most likely to benefit from early initiation of treatment. Additional strategies in smoldering MM include vaccines, such as with PVX-410, a multipeptide vaccine designed to elicit an immune response against MM cells. This vaccine targets XBP1, CD138, and CS1, which are proteins high expressed in MM cells and involved in MM pathogenesis.287 PVX-410 is currently in phase I clinical trials and in combination with lenalidomide in order to enhance of the immune response (NCT01718899).288
Patients undergoing therapy for MM should have clinical and laboratory assessments to assure both safety and efficacy of treatment. Monoclonal protein in the serum and/or urine should be measured by immunoelectrophoresis and more sensitive immunofixation techniques as well as serum FLCs. The serum FLC assay allows for diagnosis and assessment of patients previously thought to have oligosecretory or nonsecretory disease.289 A skeletal survey should be done annually, with BM examination reserved for diagnosis and time of subsequent change in clinical status, monoclonal Ig, or hemogram. It is important to remember that reduction of serum or urine M component as objective evidence of tumor response could reflect increased protein catabolism, decreased protein production, or both. Moreover, non-M protein-secreting MM clones may emerge during treatment, so that even a marked reduction in monoclonal Ig may not correlate with decrease in tumor burden.
The Bladé criteria to assess response post-transplant were developed for the European Group for Blood and Marrow Transplant (EBMT), the International BM Transplant Registry (IBMTR), and the Autologous Blood and Marrow Transplant Registry (ABMTR).290 These criteria include a more sensitive and rigorous definition of complete response (CR), including the absence of paraprotein assayed by immunofixation, and excludes transient responses. More recently, serum free light chain assays have been incorporated into response criteria291–293 and the IMWG response criteria are now universally used (Table 6). Stricter definitions of complete remission resulted in the inclusion of a category of stringent complete remission in which monoclonal plasma cells are not detectable in the marrow by immunohistochemistry or immunofluorescence and the free light-chain ratio is normal. The previously used near complete remission (only positivity by serum monoclonal immunoglobulin immunofixation) is now included in the new category VGPR and the previously used minor response category was eliminated. Limitations of the new criteria are that response calls are determined by monoclonal immunoglobulin and marrow evaluation. Dynamic changes in skeletal events readily identified by modern imaging techniques such as MRI and FDG PET CT are excluded from response assessments.
Table 6 Uniform response criteria from the international myeloma working group
| Response subcategorya | Response criteria |
| CR | Negative immunofixation of the serum and urine and disappearance of any soft-tissue plasmacytomas and <5% plasma cells in marrowb |
| sCR | CR as defined above plus |
| Normal FLC ratio and Absence of clonal cells in marrowb by immunohistochemistry or immunofluorescencec | |
| VGPR | Serum and urine M-protein detectable by immunofixation but not on electrophoresis or 90% or greater reduction in serum M-protein plus urine M-protein <100 mg/24 h |
| PR | ≥50% reduction of serum M-protein and reduction in 24-h urinary M-protein by ≥90% or to <200 mg/24 h |
| If the serum and urine M-protein are unmeasurable, ≥50% decrease in the difference between involved and uninvolved FLC levels is required in place of the M-protein criteria | |
| If serum and urine M-protein are unmeasurable, and serum-free light assay is also unmeasurable, ≥50% reduction in plasma cells is required in place of M-protein, provided baseline marrow plasma-cell percentage was ≥30% | |
| In addition to the above-listed criteria, if present at baseline, a ≥50% reduction in the size of soft-tissue plasmacytomas is also required | |
| SD | Not meeting criteria for CR, VGPR, PR, or progressive disease |
CR, complete response; FLC, free light chain; PR, partial response; sCR, stringent complete response; SD, stable disease; VGPR, very good partial response; M-protein, monoclonal protein.
a All response categories require two consecutive assessments made any time before the institution of any new therapy; complete and PR and SD categories also require no known evidence of progressive or new bone lesions if radiographic studies were performed. Radiographic studies are not required to satisfy these response requirements.
b Confirmation with repeat marrow biopsy not needed.
c Presence/absence of clonal cells is based on the κ/λ of >4 : 1 or <1 : 2. An abnormal κ/λ ratio by immunohistochemistry and/or immunofluorescence requires a minimum of 100 plasma cells for analysis.
Note: SD is not recommended for use as an indicator of response; stability of disease is best described by providing the time-to-progression estimates.
Source: Rajkumar et al. 2011.294
Sequencing-based platforms, quantitative PCR (polymerase chain reaction), and multiparametric flow cytometry are now being employed to detect minimal residual disease (MRD) in patients attaining at least a VGPR after primary therapy that may be of significant prognostic value. Martinez-Lopez et al. recently reported the results of sequencing-based BM evaluations on 133 patients in VGPR or better following primary therapy. In patients achieving a CR, the TTP (time to progression) was 131 months for MRD negative patients versus 35 months for MRD positive patients. When stratified by level of MRD, the respective TTP medians were 27 months for MRD ≥10−3, 48 months for MRD 10−3 to 10−5, and 80 months MRD <10−5 (p = 0.003–0.0001).295 Although not currently a standard of care, MRD assessment may play an important role in evaluating disease response in the future.
Initial treatment
Oral administration of MP was historically considered a standard form of therapy that produces objective response in up to 50–60% of patients.296, 297 Multiple older studies have examined whether MP is as effective as combination chemotherapy (CCT) (generally with older agents such as vincristine and cyclophosphamide). In an attempt to determine which patients, if any, do better with more aggressive therapy, Gregory et al.298 examined published reports of 18 randomized controlled trials comparing MP with CCT in the primary treatment of 3814 patients. The overall results suggested that there was no difference in efficacy between these treatment modalities. The studies with a high MP 2-year survival rate showed a survival difference in favor of MP, whereas those with a low rate suggested a difference in favor of CCT. These results imply that, rather than there being no difference between MP and CCT, MP is superior for patients with an intrinsically good prognosis and inferior for those patients with a poor prognosis.
Immunomodulatory drugs
The introduction of thalidomide, an IMiD, was the first in a series of improvements over the MP regimen. The incorporation of thalidomide in combination with MP (MPT, melphalan, prednisone, thalidomide) in newly diagnosed patients with myeloma over the age of 65 years299 resulted in a 76% complete or PR rate compared to 47% in the MP arm. This translated into a doubling of the 2-year event-free survival (EFS) to 54% versus 27%. On the basis of these data, MPT was the standard of care for transplantation-ineligible patients. However, all studies showed an increase in adverse events in the MPT arm, including infections, neuropathy, and thromboembolism, suggesting that thromboprophylaxis and antimicrobial prophylaxis are required.300 Melphalan, prednisone, and lenalidomide (MPR) is another effective regimen in this population. Palumbo et al. then went on to evaluate the efficacy and safety of induction therapy with MPR followed by lenalidomide maintenance therapy (MPR-R), as compared with MPR or MP without maintenance therapy, in patients with newly diagnosed MM who were ineligible for transplantation. At a median follow-up of 30 months, the median PFS was 31 months for MPR-R versus 14 months for MPR and 13 months for MP. This benefit was observed in patients 65–75 years of age but not older than 75. Response rates were superior for the lenalidomide-containing regimen 77% for MPR-R and 68% for MPR versus 5% with MP.301
Randomized trials with the use of other novel agents such as bortezomib with MP have proven benefits as well. For example, the VISTA trial compared the regimen of bortezomib, melphalan, and prednisone (VMP) MP in patients who were not candidates for autologous stem-cell transplant.302 OS was significantly improved in the VMP group versus MP group, with 3-year OS of 68.5% versus 54%, respectively.303
Most recently, the FIRST trial, a randomized, phase III trial compared continuous lenalidomide with low-dose dexamethasone (Rd) against lenalidomide with low-dose dexamethasone for 18 cycles (Rd18) and MPT for 12 cycles.304 Median PFS for continuous Rd was 25.5 months versus 20.7 for Rd18 and 21.2 for MPT. The OS at 4 years was 59.4% for Rd versus 55.7% for Rd18 and 51.4% for MPT. In the continuous Rd arm, the ORR was 75.1% (15.1% CR, 28.4% VGPR) versus 73.4 in Rd18 (Cr 14.2%, VGPR 28.5%) and 32.3% MPT (CR 9.3%, VGPR 18.8%). The safety profile with continuous Rd was manageable as hematologic and nonhematologic adverse events were as expected for Rd and MPT. Notably, the incidence of hematological second primary malignancies was lower with continuous Rd than MPT. In newly diagnosed transplant-ineligible patients, the FIRST trial established continuous Rd as the new standard of care. There are ongoing trials looking at three drug combinations including bortezomib, lenalidomide, and dexamethasone at reduced doses and attenuated schedules in this population as well (Table 7).
Table 7 Novel agent induction for newly diagnosed transplant-ineligible patients
| Study | Regimen | Number of patients | Median follow-up (Mo) | Median OS (Mo) | Median PFS (Mo) |
| IFM 99-06305 | MP | 196 | 51.5 | 33.2 | 17.8 |
| MPT | 125 | 51.6 | 27.5 | ||
| MEL100 | 126 | 38.3 | 19.4 | ||
| IFM 01/01306 | MPT | 113 | 47.5 | 44 | 24.1 |
| MP | 116 | 29.1 | 18.5 | ||
| MM-015305 | MPR-R | 152 | 30 | 45.2 | 31 |
| MPR | 153 | NR | 14 | ||
| MP | 154 | NR | 13 | ||
| VISTA307 | VMP | 344 | 60 | 56.4 | N/A |
| MP | 338 | 43.1 | N/A | ||
| FIRST304 | Rd | 536 | 37 | 59.4%a | 25.5 |
| Rd18 | 541 | 55.7% | 20.7 | ||
| MPT | 547 | 51.4% | 21.2 |
MP, melphalan, prednisone; MPT, melphalan, prednisone, thalidomide; MEL 100, melphalan 100 mg/m2, MPR, melphalan, prednisone, lenalidomide; MPR-R, melphalan, prednisone, lenalidomide induction followed by lenalidomide maintenance; VMP, bortezomib, melphalan, prednisone; Rd, lenalidomide, low-dose dexamethasone continuously; Rd18, lenalidomide, low-dose dexamethasone for 18 cycles; OS, overall survival; PFS, progression-free survival; NR, not reached.
a 4-year OS.
In the transplant-eligible patients, two studies combined thalidomide with dexamethasone as initial therapy for MM and achieved rapid responses in two-thirds of patients, allowing for successful harvesting of PBSCs for transplantation.308, 309 Thal/dex has been compared with VAD (vincristine, doxorubicin, dexamethasone) and with dex, as initial therapy for patients before collection of autologous stem cells and transplantation. In a case control analysis, Cavo et al.310 showed that thal/dex achieved higher overall response rates, whereas a randomized phase III (EGOG) trial showed statistically significantly higher response rates for thal/dex than dex-treated patient cohorts.311 This study provided the rationale for FDA (Food and Drug Administration) approval of this regimen for initial treatment of MM. Moreover, early studies show 91% responses, including 6% complete and 32% near complete/VGPRs to lenalidomide combined with dex.312 On the basis of these promising results, a phase III trial in the United States headed by ECOG investigated the role of len/dex in newly diagnosed MM. The study design allowed all patients to stay on-study for the first four cycles only for response assessment, after which patients could go off-study to proceed with stem-cell transplant. Safety data from this trial found that combining lenalidomide with the low-dose dexamethasone regimen was preferable to the combination with high-dose dexamethasone, with a reduction in grade 3 or higher nonhematologic adverse events (48% vs 65%), including thromboembolism (12% vs 26%), and infections (9% vs 16%) in the two treatment arms of the trial.313, 314 The low-dose dexamethasone-containing regimen did lead to an increased occurrence of grade ≥3 neutropenia (20% vs 12%). Importantly, the combination with low-dose dexamethasone had a survival benefit over combination with high-dose dexamethasone, with a 1-year OS of 96% and 87%, respectively.313, 314 Prophylaxis against clotting with aspirin, warfarin, or subcutaneous low molecular weight heparin is needed when patients are treated with lenalidomide.315, 316
Proteasome inhibitors
Richardson et al.317 examined single agent bortezomib and Jagannath et al.318 tested bortezomib combined with dex as initial therapy; in both cases, high frequency and extent of response were noted. In a phase I/II trial, Richardson et al.319 demonstrated the safety and efficacy of the combination of lenalidomide, bortezomib, and dexamethasone (RVD), which showed an unprecedented overall response rate of 100%. Building upon this work, the benefits of combination therapy with RVD as first-line therapy were seen in the results of two-phase II trials—the IFM 2008 trial and the EVOLUTION trial.320, 321 The ORR after induction in the IFM trial was 97% (13% sCR, 16% CR, and 54% ≥VGPR). The EVOLUTION trial was designed to compare RVD with CyBorD (cyclophosphamide, bortezomib, and dexamethasone) in a randomized, multicenter setting. The ORR for the RVD arm after primary treatment followed by maintenance with bortezomib for four 6-week cycles was 85% (24% CR, 51% ≥VGPR).
Carfilzomib is an epoxomicin analog second-generation proteasome inhibitor that binds to the 20S proteasome in a highly selective and irreversible manner. Given its approval in the treatment of relapsed disease, it is now being studied in the upfront setting.322 In a dose escalation study conducted by Jakubowiak et al.,323 the combination of carfilzomib, lenalidomide, and dexamethasone (CRD) has been evaluated where patients received carfilzomib (20, 27, or 36 mg/m2, days 1, 2, 8, 9, 15, and 16 for 8 cycles then days 1, 2, 15, and 16) with lenalidomide 25 mg days 1–21 and dexamethasone 40 mg weekly cycles 1–4 then 20 mg weekly cycles 5–8 in 28-day cycles. After eight cycles, patients received the regimen every other week for eight cycles. After 24 cycles, maintenance with lenalidomide was recommended off-study. After a median of 12 cycles, 62% achieved at least a near-CR and 42% an sCR. 24-Month PFS was estimated at 92%. The toxicity profile was acceptable and notable for limited peripheral neuropathy.
Doublet and especially triplet regimens of novel drugs in combination with dexamethasone can induce complete remission rates comparable to transplantation regimens,310, 324 as described. Examples of modern regimens in use include doublet combinations of lenalidomide and dexamethasone and bortezomib and dexamethasone, as well as, the triple combination of RVD, CyBorD, and CRD (Table 8).
Table 8 Novel agent induction for newly diagnosed transplant-eligible patients
| Study | Regimen | Number of patients | Cr/nCR (%) | ORR (%) | Outcome |
| Rajkumar et al.313 | RD Rd | 223 222 | 18 14 | 79 68 | OS: 87% at 1 year OS: 96% at 1 year |
| Harousseau et al.325 | VAD Bd | 121 121 | 6.4 14.8 | 62.8 78.5 | PFS 30 months PFS: 36 months |
| Reeder et al.326 | CyBorD | 33 | 39 | 88 | N/A |
| Richardson et al.319 | RVD | 66 | 39 | 100 | OS 97% at 18-months |
| Jakubowiak et al.323 | CRD | 53 | 62 | 98 | PFS 92% 24-months |
RD, lenalidomide, high-dose dexamethasone; Rd, lenalidomide, low-dose dexamethasone; VAD, vincristine, doxorubicin, dexamethasone; Bd, bortezomib, low-dose dexamethasone; CyBorD, cyclophosphamide, bortezomib, dexamethasone; RVD, lenalidomide, velcade, dexamethasone; CRD, carfilzomib, lenalidomide, dexamethasone; OS, overall survival; PFS, progression-free survival; N/A, not available.
In patients where stem-cell collection is planned, combinations that include alkylating agents such as melphalan should be avoided as damage to normal hematopoietic stem cells can be incurred, which may render it impossible to collect stem cells for auto-HSCT. Lenalidomide may also hamper the collection of stem cells, although stem cell mobilization with growth factors and chemotherapy may overcome the myelosuppressive effects of lenalidomide.327–330 The number of cycles of treatment, especially with lenalidomide-containing regimens, is limited to roughly four cycles, as additional cycles may compromise the ability to collect stem cells.327, 331
Combination therapy with novel drugs achieves complete remission rates comparable to those obtained with auto-HSCT. This has led to the design of ongoing studies that compare novel agents followed by auto-HSCT with novel agents and then auto-HSCT in case of disease relapse. Novel agents seem to be able to overcome some of the cytogenetic adverse prognostic factors such as del 13, t(4;14), and del 17p. It is too early to abandon auto-HSCT as the follow-up in clinical trials with new agents is too short to determine whether increased complete remission rates translate into durable remissions and EFS and OS. Complete remission rates as a surrogate marker for eventual outcome may prove to be inadequate. The DETERMINATION trial (NCT01208662) is an ongoing phase III, multicenter randomized trial of upfront high-dose melphalan with autologous stem-cell transplant versus transplant at relapse for myeloma patients up to age 65; all patients receive RVD as induction. This trial is designed to address this question of the role of autologous transplant, upfront or at time of relapse, in the context of novel drugs. It is likely that autologous transplant will add to the benefits noted with new drugs.
Radiation therapy
Radiation therapy for MM is used for treatment of localized disease, including plasmacytoma or spinal cord compression syndrome, and is frequently used for palliation. Hemibody radiation therapy has been utilized, either as a consolidation following induction CCT or as salvage therapy for chemotherapy-resistant MM.332, 333 Total body irradiation (TBI) has been used as a component of ablative therapy before hematopoietic stem-cell grafting but is rarely used as high-dose melphalan has equivalent efficacy and less toxicity.334
High-dose therapies
The rationale for the administration of alkylating agents (melphalan, cyclophosphamide, and busulfan) in a higher-than-conventional dose with or without TBI, followed by transplantation of syngeneic, allogeneic, and autologous BM or peripheral blood progenitor cells (PBPCs) is as follows: plasma-cell dyscrasias remain uniformly fatal; multiple studies document sensitivity of MM cells to chemotherapy and radiotherapy; and CRs can be obtained with HDT.
Autologous stem cell transplantation
High-dose chemoradiotherapy followed by transplantation of either autologous BM or PBPCs has also achieved high (40%) CR rates, but the median duration of these responses has unfortunately only been 24–36 months.335, 336 Patients with sensitive disease and who are less heavily pretreated have the most favorable outcomes. Most importantly, a national French trial of 200 patients with MM who received two courses of VMCP alternating with VBAP and were then randomized to receive either conventional chemotherapy (eight additional courses of VMCP/VBAP) or HDT (melphalan and TBI) followed by autologous BMT has demonstrated significantly higher response rates, EFS and OS for those patients treated with high dose compared to those receiving conventional therapy.337 A second randomized trial in MM examined the relative merits of HDT either early versus late as salvage therapy for relapse after conventional therapy.338 The OS was 64 months in both groups, but the quality-adjusted time without symptoms and toxicity (Q-TWIST) strongly favored the early transplant cohort. The United States Intergroup Trial supports this view.339 The IFM has conducted a randomized trial comparing high-dose melphalan at 200 mg/m2 versus melphalan at 140 mg/m2 plus TBI as ablative therapies.334 Although response rates and EFS were comparable, toxicity and OS were superior in the high-dose melphalan alone arm, suggesting that TBI should not be considered part of the ablation regimen. A Scandinavian population-based study demonstrated prolonged survival for patients with MM <60 years of age treated with intensive therapy compared with historical controls who received conventional therapy.340 An additional randomized MRC trial confirmed a 12-month survival benefit in patients receiving high dose compared to conventional therapies.341 Although these studies are encouraging, it is unlikely that patients are cured after a single high dose and stem-cell autografting regimen. In all the studies, the median EFS was prolonged; in four of five studies, transplant led to higher CR rate; and in three of five trials OS was improved (Table 9).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


