Pheochromocytoma and multiple endocrine neoplasia syndromes
GENETIC COUNSELING AND TESTING
PHEOCHROMOCYTOMA AND PARAGANGLIOMA
HEREDITARY ENDOCRINE NEOPLASIA SYNDROMES
Familial Isolated Pituitary Adenomas
Familial Paraganglioma Syndromes
Hyperparathyroidism-Jaw Tumor Syndrome
Multiple Endocrine Neoplasia 1 (MEN1)
Multiple Endocrine Neoplasia 2 (MEN2)
Multiple Endocrine Neoplasia 4 (MEN4)
OTHER TUMOR SYNDROMES ASSOCIATED WITH ENDOCRINE NEOPLASIA
Genetic counseling and testing
A diagnosis of an endocrine tumor in a child should always raise concern for an underlying hereditary condition, which can subsequently have medical, reproductive, psychological, or social consequences for the patient and family. Genetic counseling is a process of communication that promotes understanding, decision making, and coping related to the impact of genetic disease.1 It should be incorporated into all stages of care, both at diagnosis and during long-term follow-up, because patients’ counseling and information needs change over time and also because genetic testing and management recommendations are likely to evolve. Genetic testing, best exemplified in MEN2,2 is a multistep process that begins with an affected patient. MEN2 is one of few hereditary cancer syndromes for which predictive genetic testing is clearly indicated during childhood because an intervention (i.e., early thyroidectomy) can prevent future morbidity and possible mortality due to incurable metastatic MTC. In other disorders, such as MEN1 and VHL, genetic testing and early presymptomatic screening of an asymptomatic child may lead to an earlier diagnosis of disease but not disease that can be prevented by a prophylactic intervention. In all cases, despite the anticipated medical benefits afforded from an early diagnosis, genetic testing in children also has the potential for psychosocial harm: alteration of the child’s self-image and of the parents’ perception of the child, modification of the patient’s outlook on life, worry about the potential for genetic discrimination, early “medicalization” of an otherwise healthy child, changes in family relationships, and concerns regarding future reproductive issues. Online resources for genetic counseling and testing include the National Society of Genetic Counselors (www.nsgc.org), the National Cancer Institute “Cancer Genetics” website (www.cancer.gov/cancertopics/genetics), and “Gene Tests” (www.genetests.org), a publicly funded project that provides current and authoritative information on genetic disease and testing.
Pheochromocytoma and paraganglioma
Pheochromocytomas (PHEO) and paragangliomas (PGL) are uncommon neuroendocrine tumors that arise from neural crest–derived cells. PHEO (Figure 14-1) is the term used for a catecholamine-producing paraganglioma that occurs in the adrenal medulla, whereas PGL (Figure 14-2) refers to extra-adrenal tumors that arise from both sympathetic and parasympathetic paraganglia located outside the cerebrospinal axis.3–5 The term PHEO is often used interchangeably with PGL, but it is best to maintain a distinction between these two neoplasms due to underlying differences in genetics, clinical presentation, and malignant potential (Table 14-1).
TABLE 14-1
Major Disorders and Genes Associated with Pheochromocytoma and Paraganglioma
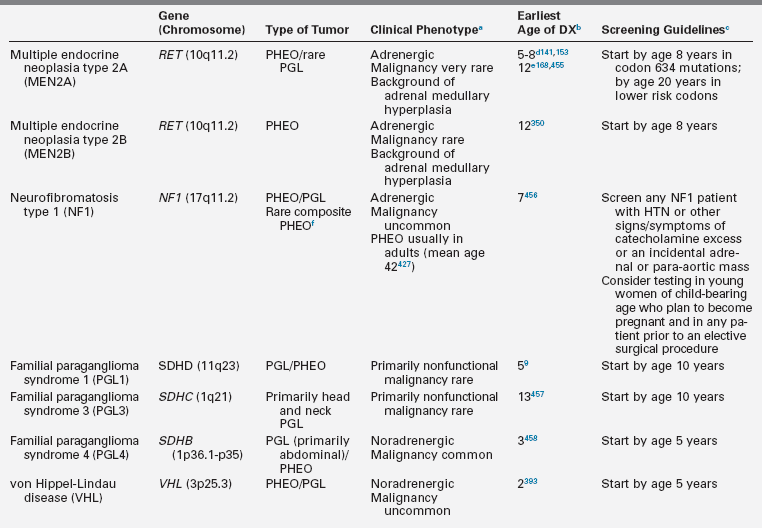
DX, diagnosis; HTN, hypertension; PGL, paraganglioma; PHEO; pheochromocytoma.
aNoradrenergic tumors almost exclusively secrete norepinephrine and normetanephrine whereas adrenergic tumors secrete epinephrine and metanephrine, in addition to norepinephrine and normetanephrine.
bEarliest age of diagnosis of PHEO/PGL and reference(s).
cIn the opinion of the authors and based on review of the literature, age at which annual screening for PHEO/PGL should be initiated for patients with a known gene mutation.
dAges of earliest PHEO onset reported in consensus guidelines via personal communication.
eAge of earliest PHEO onset published in the medical literature.
fA composite PHEO is a mixed tumor comprised of PHEO and neuroblastoma, ganglioneuroma, or ganglioneuroblastoma.
Adapted from Waguespack, S. G., Rich, T., Grubbs, E., et al. (2010). A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab, 95, 2023–2037.
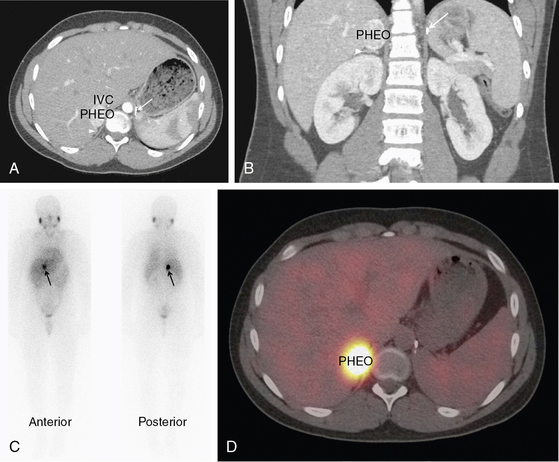
FIGURE 14-1  Pheochromocytoma. A normotensive 14-year-old male with von Hippel-Lindau disease and a history of a left pheochromocytoma and abdominal paraganglioma diagnosed at the age of 7 was found to have elevated norepinephrine and normetanephrine levels after screening with a 24-hour urine collection. Axial CT postcontrast (A) and coronal reconstruction (B) identified a vascular neoplasm (PHEO) arising from the superior right adrenal gland. IVC, inferior vena cava; arrowhead, normal adrenal gland; arrow, surgical clips from previous adrenalectomy. MIBG scan confirmed the functional nature of the tumor and ruled out other sites of disease. Planar (C) and fused SPECT/CT axial images (D) in the same patient, 24 hours after the administration of 123I MIBG. (This image can be viewed in full color online at ExpertConsult.)
Pheochromocytoma. A normotensive 14-year-old male with von Hippel-Lindau disease and a history of a left pheochromocytoma and abdominal paraganglioma diagnosed at the age of 7 was found to have elevated norepinephrine and normetanephrine levels after screening with a 24-hour urine collection. Axial CT postcontrast (A) and coronal reconstruction (B) identified a vascular neoplasm (PHEO) arising from the superior right adrenal gland. IVC, inferior vena cava; arrowhead, normal adrenal gland; arrow, surgical clips from previous adrenalectomy. MIBG scan confirmed the functional nature of the tumor and ruled out other sites of disease. Planar (C) and fused SPECT/CT axial images (D) in the same patient, 24 hours after the administration of 123I MIBG. (This image can be viewed in full color online at ExpertConsult.)
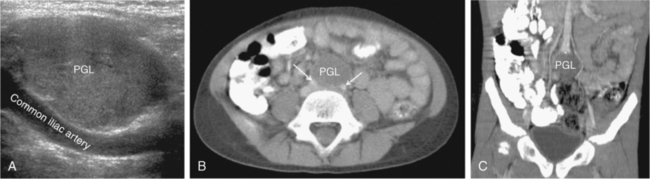
FIGURE 14-2  Paraganglioma. A 6-year-old female with a malignant paraganglioma (PGL) and an SDHB mutation presented with severe hypertension during a well-child examination. A, Abdominal ultrasound (sagittal view) revealed a homogeneous, hypervascular 3.3 cm mass at the level of the aortic bifurcation. B, Axial CT postcontrast confirmed a low attenuation tumor (arrows refer to common iliac arteries); C, Coronal reconstruction detailed the location of the neoplasm at the aortic bifurcation (*) in the organ of Zuckerkandl.
Paraganglioma. A 6-year-old female with a malignant paraganglioma (PGL) and an SDHB mutation presented with severe hypertension during a well-child examination. A, Abdominal ultrasound (sagittal view) revealed a homogeneous, hypervascular 3.3 cm mass at the level of the aortic bifurcation. B, Axial CT postcontrast confirmed a low attenuation tumor (arrows refer to common iliac arteries); C, Coronal reconstruction detailed the location of the neoplasm at the aortic bifurcation (*) in the organ of Zuckerkandl.
PHEO/PGL represent < 7% of tumors that arise from the sympathetic nervous system and have an estimated incidence of 0.3 cases/million/year or less.6,7 Up to 20% of PHEO/PGL are identified during childhood at an average age of 11 years; there is a slight predominance in boys, particularly when diagnosed under the age of 10.7–15 Less than 2% of children diagnosed with hypertension will harbor a catecholamine-producing neoplasm.16,17
All functional PHEO/PGL produce and metabolize catecholamines and contain chromaffin tissue, which refers to the brown-black color resulting from the oxidation of catecholamines after staining with chromium salts. PGL occur in all locations where paraganglia are found (from the skull base to the pelvis) and are either functional (sympathetic) or nonfunctional (parasympathetic) neoplasms, depending on the site of origin and underlying pathophysiology18–20 (see Table 14-1). PGL arising in the head and neck region are almost exclusively nonfunctional, whereas most intra-abdominal PGL (most commonly occurring within the organ of Zuckerkandl) (see Figure 14-2) are secretory tumors. The majority of these tumors diagnosed during childhood are PHEO, which synthesize and secrete catecholamines (dopamine, norepinephrine, or epinephrine) and their metabolites (including 3-methoxytyramine, normetanephrine, and metanephrine, respectively)18,21,22 (Figure 14-3). Multicentric tumors are more common in childhood presentations of PHEO/PGL.8,10,23
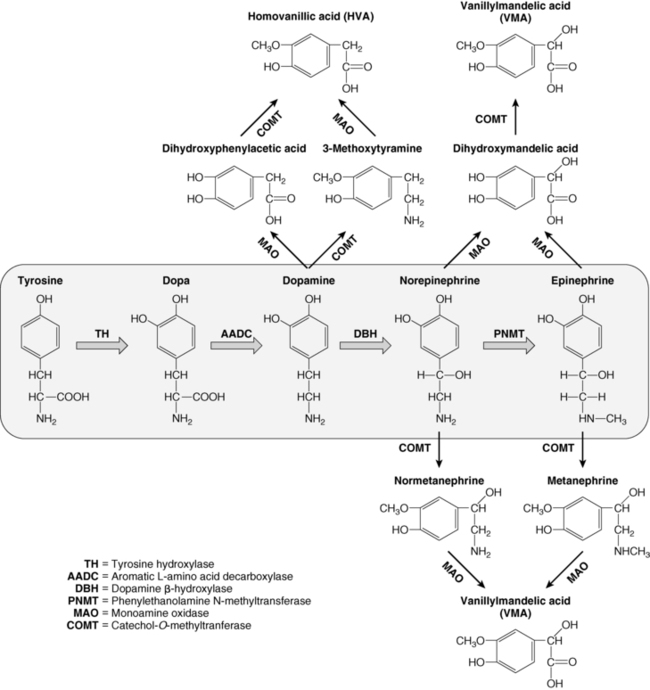
FIGURE 14-3  Catecholamine synthesis and metabolism. The catecholamines are synthesized from the amino acid tyrosine, which is converted to 3,4-dihydroxyphenylalanine (Dopa) by the enzyme tyrosine hydroxylase (TH), the rate-limiting step in catecholamine biosynthesis. Subsequent enzymatic decarboxylation (aromatic L-amino acid decarboxylase; AADC) and hydroxylation (dopamine β-hydroxylase; DBH) yields dopamine and norepinephrine, respectively, and norepinephrine is subsequently converted to epinephrine via the cytosolic enzyme phenylethanolamine N-methyltransferase (PNMT). The catecholamines are metabolized by two major enzymes: monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT).
Catecholamine synthesis and metabolism. The catecholamines are synthesized from the amino acid tyrosine, which is converted to 3,4-dihydroxyphenylalanine (Dopa) by the enzyme tyrosine hydroxylase (TH), the rate-limiting step in catecholamine biosynthesis. Subsequent enzymatic decarboxylation (aromatic L-amino acid decarboxylase; AADC) and hydroxylation (dopamine β-hydroxylase; DBH) yields dopamine and norepinephrine, respectively, and norepinephrine is subsequently converted to epinephrine via the cytosolic enzyme phenylethanolamine N-methyltransferase (PNMT). The catecholamines are metabolized by two major enzymes: monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT).
Biosynthesis and actions of catecholamines
Dopamine, norepinephrine, and epinephrine (collectively known as “catecholamines”) are chemical neurotransmitters and hormones that play important roles in the regulation of numerous physiologic processes and the development of neurologic, psychiatric, endocrine, and cardiovascular diseases.24–27 The catecholamines are composed of a catechol (1,2-dihydroxybenzene) moiety and a side-chain amine group. They are synthesized from the amino acid tyrosine, which is converted to 3,4-dihydroxyphenylalanine (dopa) by the enzyme tyrosine hydroxylase, the rate-limiting step in catecholamine biosynthesis (see Figure 14-3). Subsequent enzymatic decarboxylation and hydroxylation of dopa yields dopamine and norepinephrine, respectively, and norepinephrine is subsequently converted to epinephrine via the cytosolic enzyme phenylethanolamine N-methyltransferase (PNMT).
The catecholamines are synthesized and stored in granules within the adrenal medulla, where they are released via exostosis into the systemic circulation in response to stressful stimuli. Dopamine and norepinephrine are also produced by postganglionic neurons in the sympathetic nervous system. Epinephrine is made only in the adrenal medulla, where it represents the predominant catecholamine (∼80%) because PNMT expression is dependent on and regulated by high local concentrations of glucocorticoids (as occurs only in the adrenal medulla, surrounded by the cortisol-synthesizing cortex with a distinct concentration gradient toward the adrenal medulla).24,28,29 The effects of catecholamines are terminated via rapid reuptake into nerve terminals by the norepinephrine transporter and via metabolism by two major enzymes: monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT)18,24,27 (see Figure 14-3).
The complex actions of norepinephrine and epinephrine are mediated by the G protein–coupled α- and β-adrenergic receptors, whereas dopamine binds to a different class of G protein–coupled dopamine receptors (five distinct receptors that are divided into two families: D1-like and D2-like)24–26 (Table 14-2). The initial classification of adrenergic receptors was based on epinephrine’s ability to both excite (α-receptor) and inhibit (β–receptor) smooth muscle. Specific agonists and antagonists characterize the adrenergic receptor subtype (α1, α2, β1, β2, and β3) and can be used as therapeutic agents. The D2 receptor is the primary dopamine receptor that is targeted for drug therapy.
TABLE 14-2
Catecholamine Receptor Classification, Function, and Pharmacology

Data from Westfall, T. C, & Westfall, D. P. (2011). Neurotransmission: the autonomic and somatic motor nervous systems. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics (12th ed.) (pp. 171–218). New York: McGraw-Hill.; Westfall, T. C., & Westfall, D. P. (2011). Catecholamines and sympathomimetic drugs. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics (12th ed.) (pp. 277–333). New York: McGraw-Hill.; Sanders-Bush, E., & Hazelwood, L. (2011). 5-Hydroxytryptamine (serotonin) and dopamine. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics (12th ed.) (pp. 335–361). New York: McGraw-Hill.
Clinical presentation
The clinical presentation of pediatric PHEO/PGL is highly variable. Children with these tumors can come to attention due to symptomatic catecholamine hypersecretion, symptoms due to tumor mass effect (e.g., pain), an incidental radiographic finding, or because of screening for one of the associated hereditary tumor syndromes7,30,31 (see Table 14-1). PHEO/PGL may also arise in the setting of cyanotic congenital heart disease.32,33 Given their neuroendocrine origin, PHEO/PGL can very rarely co-secrete other hormones that result in a clinical syndrome of ectopic hormone excess, such as gigantism (growth hormone-releasing hormone), Cushing syndrome (corticotropin-releasing hormone or adrenocorticotropic hormone), hypercalcemia (parathyroid hormone–related peptide), the syndrome of inappropriate antidiuretic hormone secretion, or secretory diarrhea (vasoactive intestinal peptide).18,34
The clinical presentation of a functional PHEO/PGL depends on differences in catecholamine secretion and release as well as individual patient sensitivities to catecholamines.35 Signs and symptoms of catecholamine excess include hypertension, which is typically sustained in the majority of pediatric cases; severe headaches; paroxysmal episodes with the classic triad of headaches, palpitations, and diaphoresis (less common in children); orthostatic hypotension and syncope; pallor; tremor; or anxiety.* PHEO/PGL in children can also cause nonspecific signs and symptoms such as blurred vision; abdominal pain, diarrhea, and other gastrointestinal symptoms; weight loss; hyperglycemia; polyuria and polydipsia; low-grade fever; and behavioral problems/decline in school performance.† Bladder PGL can present with hematuria and paroxysmal symptoms during micturition.34,39
Complications of catecholamine excess can include hypertensive crisis, cardiomyopathy (takotsubo cardiomyopathy), arrhythmias, pancreatitis, severe constipation and intestinal pseudo-obstruction, stroke, seizures, and even multisystem crisis and death.19,34,36,38,40–42 Symptoms of parasympathetic PGL include hearing loss, pulsatile tinnitus, neck mass and other symptoms of mass effect such as voice hoarseness, pharyngeal fullness, and dysphagia.43
Compared with sporadic disease, PHEO/PGL identified during the course of prospective presymptomatic screening within the context of a familial disorder are smaller and less symptomatic (frequently asymptomatic) tumors.44–46 Although this type of clinical presentation is becoming more commonplace, there is currently no consensus as to how to approach such patients with small asymptomatic tumors, particularly in clinical settings where the risk for malignancy is low.
Evaluation
Biochemical diagnosis
The diagnosis of PHEO/PGL has been simplified by advances in the assays used to detect and quantify levels of catecholamines and their metabolites in blood and urine. The measurement of fractionated plasma or urine metanephrines (metanephrine and normetanephrine) is the most sensitive test (approaching 100% sensitivity) for the diagnosis of a sympathetic chromaffin tumor and should be the primary diagnostic test in the initial evaluation of suspected PHEO/PGL3,18,47–54 (Figure 14-4).
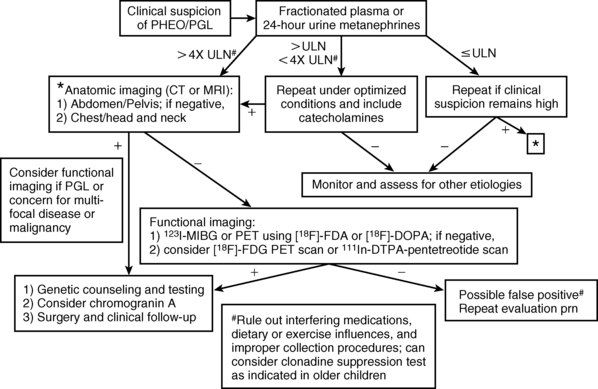
FIGURE 14-4  Diagnosis of pediatric pheochromocytoma/paraganglioma. (Adapted from Waguespack, S. G., Rich, T., Grubbs, E., et al. (2010). A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab, 95, 2023–2037.)
Diagnosis of pediatric pheochromocytoma/paraganglioma. (Adapted from Waguespack, S. G., Rich, T., Grubbs, E., et al. (2010). A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab, 95, 2023–2037.)
The high sensitivity of metanephrine testing is based on the fact that there is continuous intratumoral metabolism of catecholamines, a process that occurs independently of catecholamine release, which can occur intermittently or at low rates.50 An elevation of metanephrines greater than fourfold above the reference range is associated with almost 100% probability of the presence of a catecholamine-secreting tumor.55 Any drugs known to interfere with these assays (e.g., acetaminophen, tricyclic antidepressants, phenoxybenzamine, and decongestants, among others3) should be discontinued prior to testing. Dietary restrictions need not be routinely employed but should be considered if the assay utilized measures only deconjugated normetanephrines or if a dopamine-secreting tumor is suspected.56 The measurement of the catecholamine metabolites vanillylmandelic acid (VMA) and homovanillic acid (HVA) is no longer recommended for the evaluation of PHEO/PGL. However, testing for HVA and VMA in spot urine samples remains a critical component of the evaluation of neuroblastoma, where these analytes have a high sensitivity and specificity for tumor detection.57
In patients with mildly elevated metanephrine levels in whom a false-positive test result is suspected, consideration should be given to measuring these analytes in the supine position, 30 minutes after an indwelling needle or catheter is inserted into the vein.3,53 Clonidine suppression and glucagon stimulation tests58–60 have been a component of the diagnostic algorithm in adults but are rarely required and, in the case of the glucagon stimulation test, have been largely abandoned due to insufficient diagnostic sensitivity.61 Furthermore, these tests have not been validated in the diagnosis of childhood PHEO/PGL and are not currently recommended.
Catecholamine-producing tumors can be subclassified as being either noradrenergic or adrenergic based uon their pattern of catecholamine release.62,63 Noradrenergic PHEO/PGL secrete norepinephrine and normetanephrine, as seen in VHL disease and in tumors associated with the familial PGL syndromes.15,62,64–66 Adrenergic tumors secrete both epinephrine and norepinephrine and their metabolites, and these tumors are more commonly PHEO that arise sporadically or within the clinical context of MEN2 or NF1.50,62,64,66 This differential secretion of catecholamines is due to decreased expression of PNMT in noradrenergic tumors.64
Dopamine-secreting tumors are rare and are typically extra-adrenal SDHx-mediated paragangliomas.67–69 The measurement of plasma methoxytyramine (see Figure 14-3) may help to identify such a tumor, particularly in the context of an SDHx mutation,70 but this test is not widely available. A dopamine-secreting tumor should be considered in normotensive patients identified to have a mass that appears consistent with a PHEO/PGL, in which case dopamine and its metabolites, homovanillic acid and methoxytyramine (if available), should also be measured.68,70–72 For prospective screening in patients with an SDHx mutation, total catecholamines should also be checked in addition to metanephrines due to this possibly.
Chromogranin A is a major secretory protein present in the soluble matrix of chromaffin granules that serves as an effective tumor marker that may correlate with PHEO/PGL size and malignant potential.15,47,73–75 Chromogranin A also appears to be a useful marker in the biochemically silent, SDHB-related paraganglioma,20,69 making it a potentially useful test in the screening of asymptomatic SDHx mutation carriers.
Radiographic studies
Once a diagnosis of catecholamine excess is established from biochemical testing, radiographic studies should be undertaken to identify the location of the tumor(s)19,53 (see Figures 14-1, 14-2, and 14-4). These tests include both anatomic imaging using computed tomography (CT) or magnetic resonance imaging (MRI) and functional nuclear scintigraphy obtained concomitantly with single-photon emission computed tomography (SPECT)/CT, chiefly using radiolabeled meta-iodobenzylguanidine (MIBG).76 Initial radiographic studies should include anatomic cross-sectional imaging of the abdomen and pelvis, followed by imaging of the neck and chest if the initial studies are unrevealing18,50 (see Figure 14-4). CT and MRI have similar diagnostic sensitivities, and so the imaging test of choice is best determined by local practices and patient preference.53,77 Abdominal ultrasound may also be considered in young children, if local expertise permits (see Figure 14-2). Catecholamine-producing tumors are highly vascular (and hence enhancing) neoplasms that commonly contain necrotic, cystic, or hemorrhagic areas; on MRI, they may exhibit a classic hyperintense appearance on T2-weighted images.18,78
MIBG, a synthetic compound that bears structural similarity to norepinephrine (except it also has a guanidine side chain that resists metabolism), accumulates preferentially in adrenergic tissues, namely due to reuptake via the norepinephrine transporter system.79,80 MIBG utilized for diagnostic purposes is radiolabeled with either 123I or 131I, although 123I-MIBG is the agent of choice because of its superior imaging properties and substantially lower radiation dose.76 123I-MIBG scanning is a sensitive and specific test (94% and 92%, respectively76) that can confirm the catecholamine-producing nature of a tumor, localize tumors not seen with cross sectional imaging, and potentially identify other sites of disease, although its use is more limited in malignant disease.50,51,81–84 Whether or not MIBG scintigraphy is required in all cases where tumor is located by CT or MRI remains an area of debate,53 but it should be considered in known syndromic disease where there is a higher risk of multifocal disease. Prior to 123I-MIBG scanning, care should be taken to ensure that the patient is not taking medications (decongestants, calcium channel blockers, or labetalol) that are known to decrease MIBG uptake, and potassium iodide should be administered to block thyroid uptake of radioactive iodine.80,85 Because of the limitations of MIBG testing, other nuclear imaging modalities have been studied: somatostatin receptor scintigraphy using 111In-DTPA-pentetreotide (octreotide), [18F] fluoro-dihydroxyphenylalanine (18F-DOPA) positron emission tomography (PET), [18F]-fluorodopamine (18F-FDA) PET, or [18F] fluorodeoxyglucose (FDG) PET.50,77,86–88 Although some of these functional studies, particularly 18F-DOPA and 18F-FDA PET, are likely to be superior to scanning with MIBG,89–91 not all centers can perform these studies. [18F]FDG PET appears to be superior in the evaluation and workup of malignant PHEO/PGL, particularly in SDHB mutation carriers.91–94
Genetic issues
The majority of apparently-sporadic PHEO/PGL that present in children and young adults are due to an identifiable germline mutation in one of several tumor-predisposing genes9,15 (see Table 14-1). The most commonly associated syndrome is VHL followed by the familial paraganglioma syndromes (PGL1-4) and MEN2 (see separate sections presented later in the chapter). In all cases, synchronous or metachronous tumors can occur in both adrenal glands as well as extra-adrenal sites, underscoring the need for lifelong follow-up testing and appropriate genetic counseling and testing. Table 14-1 lists the major hereditary syndromes associated with PHEO/PGL in children. Knowledge regarding the genetic causes of catecholamine-producing tumors is rapidly expanding and genes associated with the development of PHEO/PGL include egl nine homolog 1 [C. elegans] (EGLN1 also known as PHD2)95; kinesin family member 1B (KIF1B)96; transmembrane protein 127 (TMEM127)97; succinate dehydrogenase complex, subunit A (SDHA)98; MYC associated factor X (MAX)99; and succinate dehydrogenase complex assembly factor 2 (SDHAF2), the cause of PGL2.100 Most recently, gain-of-function germline and somatic mutations in HIF-1 alpha-like factor (HIF2A; also known as EPAS1) have been associated with the development of PGL in individuals with congenital polycythemia.101,102
Family history, clinical presentation of the patient, and differences in the biochemical phenotype help to prioritize genetic testing.31,103 PHEO/PGL that arise in the context of VHL or the familial paraganglioma syndromes occur at younger ages and are typically noradrenergic tumors, producing almost exclusively norepinephrine and normetanephrine.15,66 Adrenergic tumors (which secrete epinephrine and metanephrine, in addition to norepinephrine and normetanephrine), are seen in MEN2, NF1, and sporadic cases. In general, VHL is the major gene of interest in children with PHEO and SDHB is the suspected gene in patients with PGL or malignant disease.9,15 Evaluation for RET proto-oncogene germline mutations is recommended only in the rare case of a child with an apparently sporadic PHEO who exhibits an adrenergic phenotype, because medullary thyroid carcinoma usually presents before PHEO in most individuals with MEN2.2,31 NF1 is usually clinically diagnosed and therefore testing for mutations in the NF1 gene in the context of an apparently sporadic tumor will be of very low yield and therefore is not recommended.
Management
Surgical therapy
Surgical resection is the mainstay in the treatment of PHEO/PGL. A preoperative biopsy is not indicated and potentially dangerous.104 The procedure of choice for most PHEO is laparoscopic adrenalectomy, either using transperitoneal or retroperitoneal approaches.4,105–111 Laparotomy should be contemplated in patients with large PHEO or a concern for underlying malignancy based on the clinical presentation, genetic background, or radiographic appearance of the tumor.4 In the setting of bilateral PHEO or known hereditary PHEO, a cortical-sparing procedure should be performed to minimize the risk of the lifelong glucocorticoid and mineralocorticoid replacement and the attendant risks of primary adrenal insufficiency.4,107,112–114 Because it is extremely difficult to preserve a vascularized portion of adrenal cortex sufficient to prevent corticosteroid dependence without also leaving some residual adrenal medulla, there is a risk for recurrent PHEO in the remnant; limited data suggest that recurrence rates in this setting are between 10% and 38%.10,30,112,115 The surgical approach for removal of a PGL depends on the location of the tumor but in selected cases of abdominal disease can also be performed laparoscopically.19,108 Head and neck PGL can be expectantly monitored or treated with surgery or radiation.4
It is important that the anesthesiologist have experience with the intraoperative management of PHEO/PGL, because dysrhythmias can occur and blood pressures can be quite labile.116,117 Both intravenous antihypertensive medications (esmolol, labetalol, nitroprusside, phentolamine, etc.) and vasopressors (e.g., phenylephrine and norepinephrine) should be readily available for intraoperative use. The greatest risk for hypertension occurs during anesthesia induction and manipulation of the tumor, whereas hypotension is most likely to occur after ligation of the adrenal vein, when the abrupt decline in catecholamine concentrations leads to vasodilation.4 Postoperatively, the patient should be monitored for the two major complications of hypotension and hypoglycemia.3,4,116,117 Hypertension may persist for days to weeks following surgery. In patients who have had a cortical-sparing adrenalectomy in the context of bilateral PHEO resection, stress glucocorticoids should be provided and a high-dose cosyntropin stimulation test obtained prior to hospital discharge to determine the need for adrenal steroid replacement.
Medical preparation for surgery
Once the diagnosis of a PHEO/functional PGL has been confirmed, medical therapy to normalize the blood pressure and mitigate the signs and symptoms of catecholamine excess should be initiated (Table 14-3). If surgery is planned, medical treatment should be taken for at least 1 to 2 weeks prior to surgery. This is done to minimize the potential complications that may arise from acute catecholamine surges during the induction of anesthesia and manual manipulation of the tumor.11,19,116,118 No universal algorithm exists for the medical management of a PHEO/PGL prior to surgery. Nevertheless, blockade of α-adrenergic receptors is usually the therapy of choice and effective α-receptor blockade improves symptoms, lowers blood pressure, and expands the vascular bed and blood volume. The primary agent used in children is the nonselective α-blocker phenoxybenzamine.7,18,19,116 Side effects of phenoxybenzamine can include nasal congestion, symptomatic orthostasis, and tachycardia. Due to its long half-life, it may also increase the risk of postoperative hypotension.3,11,51,60 Selective α1-blockers such as prazosin and doxazosin and calcium channel blockers such as nifedipine can also be utilized.11,60,119–121 Although labetalol and carvedilol, drugs with both α- and β-antagonist activity, are attractive options for preoperative blockade, they are not universally recommended for primary medical treatment because of their lower α-adrenergic receptor blockade relative to their β-antagonist activity.121 Metyrosine is a competitive inhibitor of tyrosine hydroxylase, the rate-limiting step of catecholamine biosynthesis (see Figure 14-3), and it can also be utilized as part of the preoperative preparative regimen.121,122 However, not all centers use metyrosine routinely due to its potential significant side effects (sedation, diarrhea, and extrapyramidal manifestations) and unclear benefit in most cases.4,18,31 Symptomatic postural hypotension may be seen at the beginning of medical therapy, particularly with large biochemically active tumors, so it is imperative to start at low doses and increase the dose or frequency every few days until the blood pressure is normal for age and height and the patient is minimally orthostatic. Phenoxybenzamine is only supplied as a single dose (10-mg capsule), so it will need to be compounded by the pharmacy to allow for the administration of the lower doses needed in younger children. Phenoxybenzamine is also an expensive drug, which makes the selective α1-blockers a more attractive option in many cases.
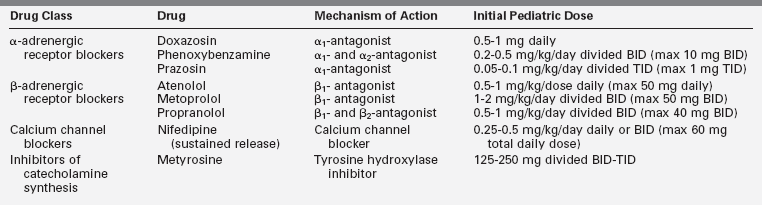
Once alpha blockade has been established, a β-blocking agent (see Table 14-3) is typically added to control reflex tachycardia.121 A β-blocker should not be used as a single agent because of the possibility of worsening symptoms and hypertension due to unopposed catecholamine effects at α-adrenergic receptors.3 A few days prior to surgery, oral salt loading (either via increased dietary intake or with sodium chloride tablets) is recommended to expand the blood volume in order to mitigate postoperative hypotension. Some centers also routinely admit patients for intravenous fluids prior to PHEO/PGL resection,121 and this should be considered for very symptomatic children with large tumors.
Prognosis and follow-up
The prognosis of children diagnosed with PHEO/PGL is excellent with 5- and 10-year survival rates of 98% and a 20-year survival rate of 84%.15 Based on data from a British tumor registry, the incidence of malignant disease in children is estimated to be 0.02 per million per year.7 Approximately 12% of pediatric PHEO/PGL are malignant,8 although some referral centers report a malignancy rate in tumors diagnosed during childhood as high as 65%.14,15,30 The high rate of malignancy from some studies may in part reflect a referral bias. On the other hand, the latency period between diagnosis and confirmation of metastatic disease is 9 years on average15; thus, the true rate of malignancy may be higher than previously recognized because it can only be identified during long-term systematic follow-up of patients diagnosed with PHEO/PGL during childhood. Children with metastatic disease typically demonstrate a more indolent clinical course with average overall survival > 6 years after the diagnosis of metastatic disease.15
There is no single histologic feature or immunohistochemical profile that is independently able to predict metastatic potential in a resected PHEO/PGL, but features noted more frequently in malignant tumors include extra-adrenal location, confluent tumor necrosis, absence of hyaline globules, coarse nodularity of the primary tumor, high proliferative index, and size greater than 5 cm, among others.22,123–125 Malignancy is therefore only established by the identification of distant metastases in a site where paraganglia are not normally located (primarily bones but also lymph nodes, liver, or lungs).15,22,123 The risk of malignant disease is greater for extra-adrenal sympathetic PGL than for PHEO or nonsecretory head and neck PGL, and overall prognosis is worse for these patients.15,125 The highest risk for malignancy and death is in SDHB-related sympathetic PGL, which represents 50% or more of malignant tumors.15,65,69,126–130
Because PHEO/PGL can have unpredictable behavior and because children are at risk for the development of metachronous primary tumors, delayed metastasis from previously treated neoplasms, and local recurrence (in the case of cortical-sparing procedures), long-term follow-up with biochemical screening and intermittent imaging studies is required, particularly for children with a PGL or a known SDHB mutation.3,7,10,15,19,22,30,112,115 For asymptomatic children with an identified genetic mutation predisposing them to the development of a PHEO/PGL, annual biochemical screening is advised, with the age of initial screening determined by the specific gene mutation (see Table 14-1). Furthermore, occasional cross-sectional imaging, typically MRI because of the lack of radiation exposure, is recommended periodically for follow-up of patients at high risk of recurrence or malignant disease (e.g., abdominal paragangliomas) or at risk for developing a PHEO/PGL that may not be identified on biochemical testing alone (e.g., familial paraganglioma syndromes), although the optimal screening strategy has yet to be determined.131
Medullary thyroid carcinoma
MTC is a malignant neuroendocrine tumor that arises from the neural crest–derived, calcitonin-producing parafollicular C cells of the thyroid gland.132,133 It comprises only a small minority of thyroid malignancies diagnosed in patients under the age of 21 years, although it is the most common thyroid malignancy diagnosed at less than age 5 years.134 The overall age-adjusted incidence of MTC during childhood is < 0.5 cases per million per year, with a fairly equal female:male ratio, unlike the differentiated thyroid carcinomas, which are more frequent in girls than boys.134–136
When diagnosed during childhood, MTC primarily results from a dominantly inherited or de novo activating mutation in the multiple endocrine neoplasia (MEN) type 2A or type 2B or familial MTC (FMTC). FMTC is more commonly being recognized as a phenotypic variant of MEN2A with decreased penetrance or delayed onset of the other neoplastic manifestations.141,142 RET is a member of the cadherin superfamily and encodes a receptor tyrosine kinase that has an extracellular binding domain and an intracellular tyrosine kinase (Figure 14-5). The endogenous ligands (which activate RET via a high-affinity ligand-binding coreceptor, GFRα) are members of the glial cell–derived neurotrophic factor (GDNF) family, which are involved in the regulation of neural tissue development.143 The RET protein therefore plays a crucial role in the development of neural crest–derived cells, the urogenital system, and the central and peripheral nervous systems, notably the enteric nervous system.144,145
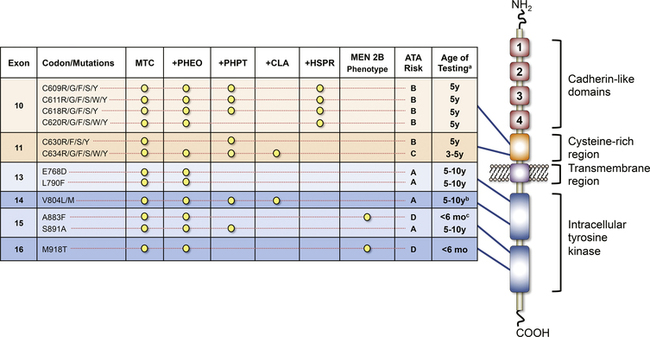
FIGURE 14-5  The RET receptor and commonly mutated codons and associated phenotypes in the MEN2 syndromes, including the recent ATA risk stratification. aAge of testing refers to the age at which clinical testing with thyroid ultrasound and basal calcitonin levels would be appropriate. It is not meant to indicate the age of testing for the presence of a RET mutation, which may be done earlier after appropriate genetic counseling, or the absolute age for early thyroidectomy. bAlthough a case of very aggressive MTC in a child diagnosed at age 6 years has been reported, an older age of disease onset is generally observed with this RET mutation. cOnly rare cases of MEN2B secondary to the A8883F mutation have been published, and the MTC phenotype of this mutation remains largely unknown. MTC, medullary thyroid carcinoma; PHEO, pheochromocytoma; PHPT, primary hyperparathyroidism; CLA, cutaneous lichen amyloidosis; HSCR, Hirschsprung disease. (This image can be viewed in full color online at ExpertConsult.)
The RET receptor and commonly mutated codons and associated phenotypes in the MEN2 syndromes, including the recent ATA risk stratification. aAge of testing refers to the age at which clinical testing with thyroid ultrasound and basal calcitonin levels would be appropriate. It is not meant to indicate the age of testing for the presence of a RET mutation, which may be done earlier after appropriate genetic counseling, or the absolute age for early thyroidectomy. bAlthough a case of very aggressive MTC in a child diagnosed at age 6 years has been reported, an older age of disease onset is generally observed with this RET mutation. cOnly rare cases of MEN2B secondary to the A8883F mutation have been published, and the MTC phenotype of this mutation remains largely unknown. MTC, medullary thyroid carcinoma; PHEO, pheochromocytoma; PHPT, primary hyperparathyroidism; CLA, cutaneous lichen amyloidosis; HSCR, Hirschsprung disease. (This image can be viewed in full color online at ExpertConsult.)  (From Waguespack, S. G., Rich, T. A., Perrier, N. D., et al. (2011). Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol, 7, 596–607.)
(From Waguespack, S. G., Rich, T. A., Perrier, N. D., et al. (2011). Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol, 7, 596–607.)
Although sporadic nonheritable tumors account for up to 75% of adult cases of MTC,141 such tumors are rare in children. In contrast to sporadic MTC, hereditary MTC is typically multifocal, bilateral, and located in the middle to upper regions of the thyroid lobes (Figure 14-6), an area where C cells are the most highly concentrated.133,146,147 Microscopic examination and calcitonin staining of the thyroid will often identify C-cell hyperplasia, which is the initial stage in an oncologic cascade that leads to the development of microscopic noninvasive MTC and ultimately lymph node and distant metastatic disease due to frankly invasive carcinoma (Figure 14-6).133,148 Patients with hereditary MTC have an age-related progression of malignant disease, with lymph node and distant metastases typically occurring years after the onset of C-cell hyperplasia.2,148 The cervical and mediastinal lymph node basins are the most common sites of metastatic disease, whereas distant sites for MTC spread typically include the lungs, liver, and bone or bone marrow. Positive lymph node status and higher stage at diagnosis predict lower disease-free survival and higher mortality.136,149–151
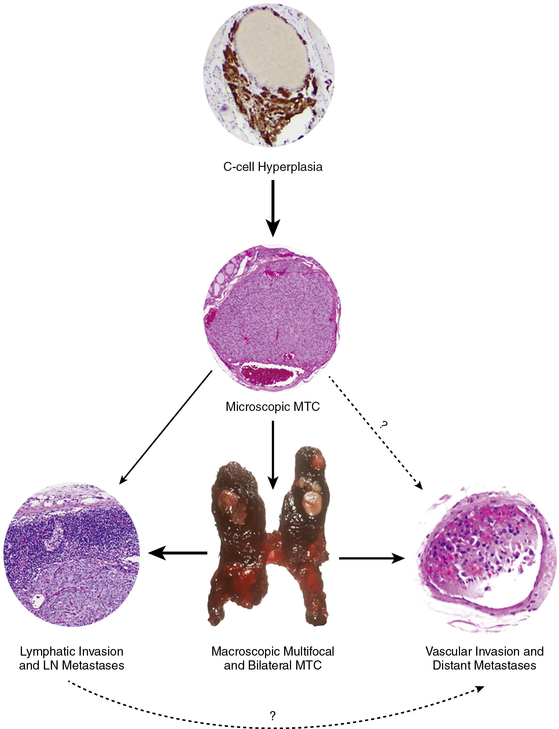
FIGURE 14-6  The development and progression of hereditary MTC in childhood. C-cell hyperplasia is the initial stage in an oncologic cascade that ultimately leads to the development of microscopic noninvasive MTC and ultimately lymph node and distant metastatic disease due to frankly invasive carcinoma. The weight of the arrows denotes the hypothetical probability of the event occurring in the typical pediatric patient with a RET codon 634 mutation. LN, lymph node; MTC, medullary thyroid carcinoma. (This image can be viewed in full color online at ExpertConsult.)
The development and progression of hereditary MTC in childhood. C-cell hyperplasia is the initial stage in an oncologic cascade that ultimately leads to the development of microscopic noninvasive MTC and ultimately lymph node and distant metastatic disease due to frankly invasive carcinoma. The weight of the arrows denotes the hypothetical probability of the event occurring in the typical pediatric patient with a RET codon 634 mutation. LN, lymph node; MTC, medullary thyroid carcinoma. (This image can be viewed in full color online at ExpertConsult.)  (From Waguespack, S. G., Rich, T. A., Perrier, N. D., et al. (2011). Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol, 7, 596–607.)
(From Waguespack, S. G., Rich, T. A., Perrier, N. D., et al. (2011). Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol, 7, 596–607.)
Clinical presentation
Children with sporadic MTC present similar to other thyroid malignancies, typically with a palpable thyroid nodule or cervical lymphadenopathy. They do not have the characteristic facial or other features found in MEN2B as described later. On the other hand, children with hereditary disease due to a RET mutation rarely present with overt clinical disease, outside of the newly identified MEN2A kindred or MTC associated with MEN2B, a diagnosis that remains uniformly delayed.152 Therefore, in the 21st century, the predominant clinical presentation of MTC during childhood is one of the presymptomatic identification of a positive RET mutation and the ultimate identification of microscopic MTC after early thyroidectomy.
Evaluation and management
Several guidelines and review papers are available to assist the clinician in the specific evaluation and management of the child with suspected or proven MTC.2,141,153–156 As with all pediatric thyroid malignancies, surgery is the cornerstone of therapy. Given the potential higher complication rates in children than adults,157 children should be operated on by a surgeon skilled in thyroid surgery and with “high volume” experience for this procedure.158 Apart from the decision of when to intervene, the thyroid surgeon must also consider the patient’s RET genotype and clinical data and incorporate this knowledge into the decision-making process to determine the optimal surgical approach.2 Meticulous and safe removal of all thyroid tissue, including the posterior capsule, is the goal of the prophylactic thyroidectomy.154 Routine central compartment (level VI) neck dissection159 is not performed in the setting of a purely prophylactic thyroidectomy because lymph node metastases are quite rare in that setting.141,148–150,154,160,161 However, if the operation is for a clinically evident hereditary tumor or for sporadic MTC, thyroidectomy and a concomitant central neck dissection should be performed. Dissection of the lateral cervical lymph node compartments (levels IIA to V) is generally performed only in cases where there is clinical evidence of lateral neck involvement.
Fortunately, life-threatening MTC rarely occurs during childhood and typically only in the clinical context of MEN2B. MTC is not sensitive to standard cytotoxic chemotherapy, which historically has incorporated the agent dacarbazine.142 Targeted molecular therapies that inhibit RET and other receptor tyrosine kinases known to be involved in angiogenesis have shown great promise in the treatment of metastatic MTC.162–164 The drug vandetanib became the first such agent approved by the U.S. Food and Drug Administration (FDA) for the treatment of adults with MTC, and preliminary results from a phase I/II trial in children with MTC have been encouraging.165
Prognosis
The prognosis of MTC diagnosed during childhood is generally excellent, with 5-year and 15-year survival rates of 95% and 86%, respectively.134 Mean survival after diagnosis is 28.3 years and the presence of distant disease at diagnosis, compared with locoregional disease, portends a worse prognosis.134,166,167 As the tumor stage increases, the risk of both locoregional and distant metastatic disease rises,136,166 although lymph node metastases may still occur when the tumor is < 1 cm in size.168–171 Many children who present with clinical MTC already have metastatic disease at diagnosis. Consequently, the majority of cases of childhood MTC that are not diagnosed before lymph node metastases occur represent incurable, albeit indolent, cancers. The aggressiveness of the clinical course can be predicted by the presence of certain RET mutations (see MEN2 section), the child’s clinical presentation, and the use of calcitonin and carcinoembryonic antigen (CEA) as tumor markers. The loss of calcitonin expression, a CEA level out of proportion to calcitonin, and a rapid CEA or calcitonin doubling time are all harbingers of an aggressive disease course.141,151,172,173
Hereditary endocrine neoplasia syndromes
Carney complex
The Carney complex (CNC) is a genetically and clinically heterogeneous disorder with autosomal dominant inheritance first described in 1985.174 It is characterized by spotty skin pigmentation affecting the lips, conjunctiva, and other mucosal surfaces (Figure 14-7); myxomas of the breast, heart, and skin; endocrine tumors or overactivity, classically primary pigmented nodular adrenocortical disease (PPNAD)175 and growth hormone/prolactin hypersecretion; and psammomatous melanotic schwannoma, among other clinical manifestations.174,176,177 Two or more distinct genetic loci are associated with CNC: the gene PRKAR1A178,179 on chromosome 17q23-q24 and an unknown gene on chromosome 2p16.180 PRKAR1A encodes for the RI-α subunit of protein kinase A, the major mediator of intracellular cAMP signaling. Most PRKAR1A mutations are inactivating point mutations, and about 30% of CNC patients have de novo disease.176,181 In patients with CNC, the overall PRKAR1A mutation detection rate is 62%.181
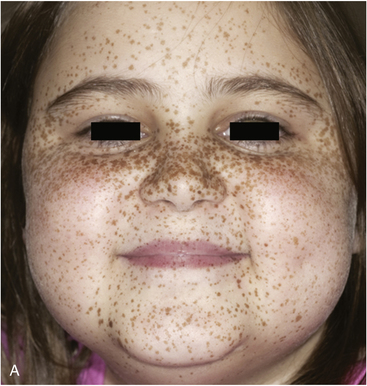
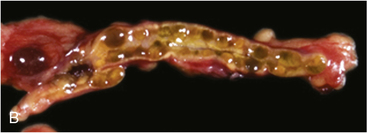
FIGURE 14-7  Carney complex and primary pigmented nodular adrenocortical disease (PPNAD). A, A 10-year-old female with ACTH-independent Cushing syndrome and the Carney complex demonstrates facial rounding, plethora, and the classic distribution of facial lentigines. B, Gross pathologic specimen of the right adrenal gland in another patient with PPNAD and the Carney complex. (This image can be viewed in full color online at ExpertConsult.)
Carney complex and primary pigmented nodular adrenocortical disease (PPNAD). A, A 10-year-old female with ACTH-independent Cushing syndrome and the Carney complex demonstrates facial rounding, plethora, and the classic distribution of facial lentigines. B, Gross pathologic specimen of the right adrenal gland in another patient with PPNAD and the Carney complex. (This image can be viewed in full color online at ExpertConsult.)
The major endocrine phenotype of the CNC is PPNAD, occurring either alone or with other CNC manifestations, in 60% of cases.181 PPNAD is a rare form of ACTH-independent hypercortisolism pathologically associated with multiple small (< 1 cm) black or brown nodules (containing lipofuscin) in an otherwise atrophic cortex175,182,183 (see Figure 14-7). The median age of onset is 34 years (with about 20% of cases occurring during childhood), and there is a female predilection after puberty.181 The hypercortisolism is typically insidious in its onset, and a classic finding in PPNAD-related Cushing syndrome is the paradoxical increase in glucocorticoid excretion during high-dose dexamethasone administration for the Liddle test.184 Adrenal imaging is of limited value for the confirmation of PPNAD, but it can suggest diffuse micronodular disease on high-resolution studies.182,183,185 The treatment of symptomatic PPNAD is bilateral adrenalectomy.
Growth-hormone hypersecretion (causing gigantism or acromegaly, depending on the age of onset) is the primary pituitary phenotype of the CNC, occurring in 10% to 12% of CNC patients.174,181,186 Pathologically, this may be associated with an adenoma or hyperplasia.186,187 Concomitant hyperprolactinemia is prevalent and is similarly caused by underlying hyperplasia or frank adenomas in rare cases.187,188 Subtle abnormalities in growth hormone and prolactin secretion may be identified in up to 75% of patients with CNC.186 Thyroid neoplasia occurs in 25% of CNC patients and differentiated thyroid carcinoma (both papillary and follicular carcinomas) have been identified in 2.5% of cases.181 Males with CNC are at risk for testicular tumors, primarily bilateral large-cell calcifying Sertoli cell tumors (LCCSCT), clinically identified in 33% to 41% of patients.181,186 Similar to the Peutz-Jeghers syndrome (discussed later), gynecomastia or palpable mass(es) may be a clinical presentation of LCCSCT, which can also be malignant in rare cases.186,189
Once CNC is diagnosed clinically or an asymptomatic child is found to harbor a germline PRKAR1A mutation, prospective monitoring for the development of the tumors and endocrine disorders characteristic of the syndrome can be undertaken,176,186 although optimal screening strategies remain largely unknown.
Familial isolated pituitary adenomas
Hereditary pituitary adenomas can occur within families outside of the context of MEN1 and other autosomal dominant, heritable endocrine neoplasia syndromes. The term familial isolated pituitary adenomas (FIPA) was first proposed in 2006.190 Pituitary tumors occurring within FIPA families clinically present similar to sporadic pituitary adenomas and all hormonal subtypes are represented. The disorder was ultimately identified in a subset of FIPA kindreds to be caused by germline mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene.191,192
Mutations in AIP represent about 15% of FIPA cases (50% of kindreds with familial somatotropinomas), and more than 50 AIP mutations have been described to date.192,193 Penetrance is incomplete and is estimated to be around 30%.193 Median age of diagnosis is 23 years (range 8 to 74 years) and half of cases present during childhood or adolescence.194 In patients < age 18 years presenting with an apparently-sporadic pituitary macroadenoma, 20% may harbor a germline AIP mutation.195 FIPA individuals with AIP mutations have tumors that are larger (overwhelmingly macroadenomas) and diagnosed at a much younger age compared with AIP-negative patients.192,194 The majority of AIP-mutated adenomas are somatotroph or mammosomatotroph adenomas, which typically present with gigantism in males.192,194 These AIP-mutated growth-hormone secreting tumors are more recalcitrant to the typical therapies prescribed for gigantism/acromegaly.194
Given the identification of AIP-related FIPA, there are no consensus guidelines as to the timing of genetic testing for AIP mutations in sporadic pituitary adenoma cases or at-risk members of an affected kindred. Recommendations for prospective clinical screening in asymptomatic AIP-mutation carriers are also not well established. However, some authors have proposed an approach to care of such patients that includes an annual hormonal and auxologic evaluation.193,196
Familial paraganglioma syndromes
The familial paraganglioma syndromes, also referred to as the familial paraganglioma and pheochromocytoma syndromes (FPPS),131 are characterized primarily by the development of parasympathetic and sympathetic PGL and PHEO.197 Four autosomal dominantly inherited FPPS syndromes (PGL1-4) have been described (see Table 14-1) and are due to germline mutations in discrete genes encoding either the subunits of the succinate dehydrogenase (SDH) enzyme gene (SDHB, SDHC, and SDHB) or a protein necessary for the flavination of SDHA: PGL1(SDHD),198 PGL2(SDHAF2),100 PGL3 (SDHC),199 and PGL4 (SDHB).200 The SDHx genes constitute the subunits for complex II of the mitochondrial respiratory chain that, when mutated, lead to stabilization of hypoxia inducible factor 1 with a subsequent state of pseudohypoxia.131 The clinical phenotype differs among the four syndromes and remains poorly characterized in some cases, such as PGL2. As an example, mutations in SDHC primarily cause benign parasympathetic head and neck PGL (also referred to as “glomus tumors” or “chemodectomas”), whereas mutations in SDHB are associated more with abdominal PGL and a high risk of malignancy (see Table 14-1, Figure 14-2). In all cases, penetrance of the clinical phenotype is less than 100% and increases with age.127,197,201,202 Mutations in SDHD (and possibly also SDHAF2) demonstrate parent-of-origin effects, with disease generally occurring only when the mutation is inherited from the father.197,203 SDHx-related tumors are associated with gastrointestinal stromal tumors (GIST), a finding that has also been referred to as the Carney-Stratakis syndrome or dyad.204,205 Other non-paraganglial malignancies described (chiefly in patients with SDHB mutations) include renal cell carcinoma and papillary thyroid carcinoma.201,206,207 A patient with an SDHD mutation was identified to have a growth hormone-secreting pituitary macroadenoma, suggesting a possible role of SDH defects and pituitary tumorigenesis.208
Hyperparathyroidism-jaw tumor syndrome
The hyperparathyroidism-jaw tumor (HPT-JT) syndrome is a heritable disorder primarily characterized by parathyroid neoplasia and primary hyperparathyroidism associated with ossifying fibromas of the maxilla or mandible. First reported in 2002, CDC73 (formerly known as HRPT2) is the only known gene in which mutations cause HPT-JT, and CDC73 mutations are identified in about 60% of HPT-JT syndrome kindreds.209 Primary hyperparathyroidism due to a single benign parathyroid adenoma is the most common clinical manifestation, but parathyroid carcinomas are quite prevalent in the syndrome, representing up to 15% of HPT cases.210 The youngest age of diagnosis of HPT has been seven years.211 Ossifying fibromas, also known as cemento-ossifying fibromas, can be quite aggressive and occur in 30% to 40% of individuals with HPT-JT syndrome.210 They are chiefly treated with surgical extirpation. Other clinical manifestations of the HPT-JT syndrome include renal lesions (cysts, hamartomas, and nephroblastoma) and uterine tumors.212,213 Prospective clinical monitoring of individuals with a CDC73 mutation includes periodic biochemistries, dental imaging, and ultrasonography of the kidney and uterus.210
Multiple endocrine neoplasia 1 (men1)
MEN1 is an autosomal dominantly inherited tumor syndrome characterized by the occurrence of glandular hyperplasia and benign or malignant neoplasia in two or more specific endocrine glands, chiefly the parathyroids, pituitary, and neuroendocrine cells of the duodenum and pancreas153,214–217 (Table 14-4). The tumors can be hormonally active or inactive and multifocality is common, except for pituitary tumors. Some patients also develop adrenocortical adenomas, carcinoid tumors, benign tumors of the skin (angiofibromas, collagenomas, lipomas), central nervous system (CNS) tumors (meningiomas and ependymomas), or uterine leiomyomas.216,217
TABLE 14-4
Characteristic Tumors and Associated Abnormalities in MEN1
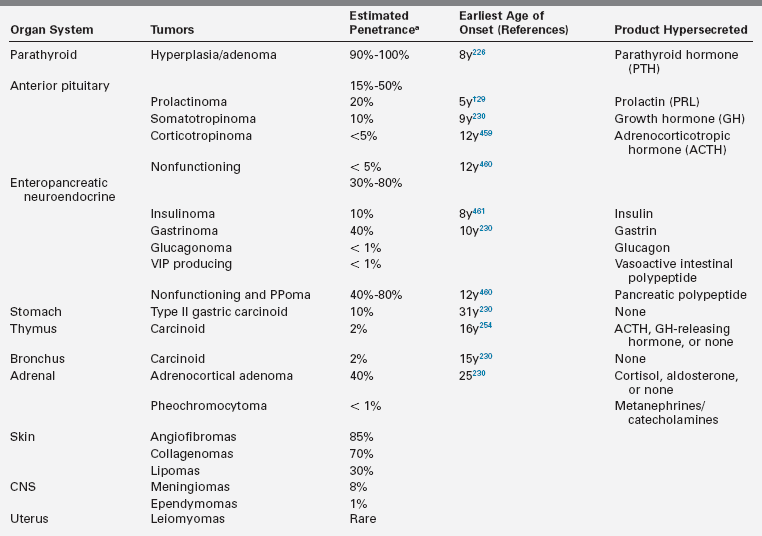
* Percentage of patients that eventually develop these clinical manifestations.153,216,217,273
†This patient had a mammosomatotroph tumor, co-secreting prolactin, and growth hormone.
The hereditary nature of MEN1 was first recognized by Wermer in 1954.218 Genetic linkage analysis among families with a clinical diagnosis of MEN1 initially localized the inherited abnormality to chromosome 11q13,219,220 which ultimately led to the identification in 1997 of germline mutations in several families with MEN1.221,222 The MEN1 gene is a tumor suppressor gene that encodes a nuclear protein (menin), which in turn plays a role in transcriptional regulation, genome stability, cell division, and proliferation.215,223,224 Advances in genetic testing have made it possible to establish a specific molecular diagnosis and screen unaffected relatives. The challenge for pediatric physicians, other caregivers, and parents is determining when to perform appropriate clinical surveillance and intervention for patients known to harbor an MEN1 gene mutation.
Epidemiology and pathogenesis
The prevalence of MEN1 is estimated at 1 to 10 per 100,000 individuals.225,226 The “two-hit” mechanism of disease, first described by Knudson in hereditary retinoblastoma,227 is illustrated in Figure 14-8. Patients with MEN1 carry one wild-type and one inactive mutant allele of the MEN1 gene (a germline mutation) in all cells. This in itself is insufficient to induce tumor formation. Subsequently, a somatic mutation (the “second hit”) in a single cell deletes the only normally functioning MEN1 gene, leads to a loss of heterozygosity at the MEN1 locus in tumor DNA, and attenuates the ordinary constraints on cell growth by menin. Thus, tumor formation is initiated from a single clone of cells.
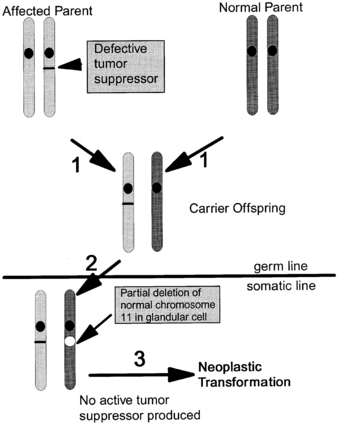
FIGURE 14-8  “Two-hit hypothesis” of tumorigenesis in MEN1.1 An affected parent passes a mutated MEN1 gene to an offspring, who inherits a normal gene from the other parent.2 Loss of heterozygosity in a somatic cell (a “second hit” that typically occurs via a subchromosomal rearrangement or deletion of the entire chromosome) deletes the remaining normal MEN1 gene.3 The absence of menin tumor suppressor activity in a cell leads to tumor formation.
“Two-hit hypothesis” of tumorigenesis in MEN1.1 An affected parent passes a mutated MEN1 gene to an offspring, who inherits a normal gene from the other parent.2 Loss of heterozygosity in a somatic cell (a “second hit” that typically occurs via a subchromosomal rearrangement or deletion of the entire chromosome) deletes the remaining normal MEN1 gene.3 The absence of menin tumor suppressor activity in a cell leads to tumor formation.
Most patients with MEN1 inherit the mutant allele from an affected parent, but about 10% of individuals with MEN1 represent de novo mutations.222,224,228 More than 90% of tumors from MEN1 patients have loss of heterozygosity due to a subchromosomal rearrangement or deletion of the entire chromosome; other mechanisms for the second hit include point mutations, small deletions or insertions within the MEN1 gene.214,215,224 There have been more than 1100 germline mutations of the MEN1 gene characterized thus far, and these mutations occur via multiple mechanisms and are distributed throughout the MEN1 gene.214,215,222,224,228 Testing by direct DNA sequencing identifies most MEN1 mutations, but approximately 5% to 10% of people with MEN1 do not have a mutation in the coding region or splicing sites of MEN1.217,224 Such patients may represent a phenocopy (see the section on MEN4), or they may harbor a mutation in untranslated regions or introns or a large gene deletion, which requires other technologies such as multiplex ligation-dependent probe amplification (MLPA) to detect.229
Clinical presentation and management
The clinical presentation of MEN1 is highly variable, even among members of the same kindred, and will depend on the location and functionality of the underlying tumor(s). Not surprisingly, functional tumors typically present 5 to 10 years earlier than nonsecretory neoplasms.230 There is almost complete penetrance of the phenotype, so MEN1 clinical and biochemical manifestations will generally develop in 80% and > 98% of patients, respectively, by the fifth decade of life.153,228,230 An age-related progression of the various endocrine tumors exists in MEN1.228,230 Penetrance of the phenotype in children with MEN1 is estimated to be < 1% before age 5 years, 7% by age 10 years, 28% by age 15, and 52% by age 20 years228 (Figure 14-9). Prospective screening of asymptomatic MEN1 mutation carriers leads to an earlier age of diagnosis,228,230–235 and thus penetrance rates are likely to increase as clinical practice evolves. In contrast to MEN2, as described in the subsequent section, there are no clear genotype-phenotype correlations in MEN1, so a physician cannot rely on the specific mutation or family history to predict the age of onset, severity, or type of an MEN1 manifestation.215,228,230
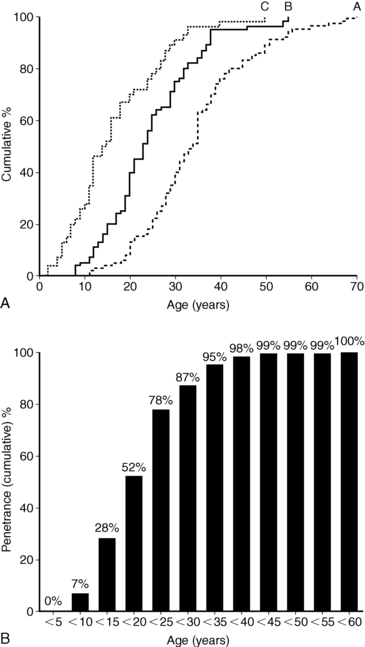
FIGURE 14-9  Penetrance of the MEN1 phenotype. Age distributions (A) and age-related penetrance (B) of MEN1 clinical manifestations in 201 carriers of a mutated MEN1 gene. Group A patients presented with clinical symptoms at the age depicted. Group B patients were asymptomatic but had positive biochemical screening at the age depicted. Group C patients were asymptomatic with negative biochemical screening with the age of their last biochemical testing shown. Groups B and C were significantly younger than Group A patients (P < .001). The age-related penetrance (B) is the proportion of mutation carriers who have developed a clinical manifestation by a given age. (From Bassett, J. H., Forbes, S. A., Pannett, A. A., et al. (1998). Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet, 62, 232–244.)
Penetrance of the MEN1 phenotype. Age distributions (A) and age-related penetrance (B) of MEN1 clinical manifestations in 201 carriers of a mutated MEN1 gene. Group A patients presented with clinical symptoms at the age depicted. Group B patients were asymptomatic but had positive biochemical screening at the age depicted. Group C patients were asymptomatic with negative biochemical screening with the age of their last biochemical testing shown. Groups B and C were significantly younger than Group A patients (P < .001). The age-related penetrance (B) is the proportion of mutation carriers who have developed a clinical manifestation by a given age. (From Bassett, J. H., Forbes, S. A., Pannett, A. A., et al. (1998). Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet, 62, 232–244.)
In the absence of treatment, MEN1-associated endocrine tumors are associated with higher mortality (50% probability of death by the age of 50 years), and the cause of death in 50% to 70% of patients with MEN1 is usually a malignant tumor process or sequelae of the disease.216,236,237 In the current era, when medical management can effectively treat symptomatic gastrinoma, mortality due to the Zollinger-Ellison syndrome has declined. Currently, the greatest risk of mortality in MEN1 is chiefly from malignant enteropancreatic tumors and thymic carcinoids.216,230,236
The specific treatment for each type of MEN1-associated endocrine neoplasm is generally similar to that for the respective sporadic tumors occurring in patients without MEN1. This section will therefore focus more on the unique aspects of these disorders as they relate to MEN1 patients. As with most hereditary tumor syndromes, patients with MEN1 and their families should be managed by a multidisciplinary team consisting of relevant specialists with experience in the management of endocrine tumors.216
Primary hyperparathyroidism
Primary hyperparathyroidism (PHPT) is the most common and earliest endocrine manifestation of MEN1.216,226,230 MEN1 patients usually have multigland hyperplasia rather than single-gland adenomas. Symptoms of PHPT are due to the underlying hypercalcemia and can be nonspecific in children, including polyuria, difficulty concentrating, fatigue, headache, poor appetite, weight loss, abdominal pain, constipation, nausea, or emesis. Similar to adults, children can have end-organ damage (nephrolithiasis, nephrocalcinosis, acute pancreatitis, and bone involvement). Children with PHPT have been shown to have more severe symptoms, with 70% to 90% symptomatic compared with 20% to 50% of their adult counterparts.238–240 Prolonged exposure to elevated PTH levels increases osteoclast activity, which can cause demineralization of bone, fractures, and eventually osteitis fibrosa cystica, the most severe skeletal manifestation of PHPT.241 Compared with patients who have sporadic PHPT, patients with MEN1 have lower bone density, a 1:1 male:female ratio (versus 1:3), lower PTH levels, and earlier age of onset (average age 20 to 25 years versus 55 years).153,242,243 The youngest reported age of MEN1-associated PHPT is 8 years.216
The diagnosis of PHPT is based on an elevated calcium level in the setting of an inappropriately normal or frankly elevated PTH level. Because all parathyroid glands may be affected in MEN1 patients, preoperative imaging is generally not utilized.244 However, in the case of reoperation, localization studies can be helpful to the surgeon.245,246 Imaging modalities primarily include ultrasound, nuclear scintigraphy using technetium (99mTc) sestamibi, and CT/MRI.247,248 Technetium (99mTc) sestamibi scans have a sensitivity of 85% to 100% in detecting single adenomas.246 Sestamibi-SPECT imaging has a sensitivity of 90% for single adenomas, but only 55% for hyperplastic glands.249 CT (sensitivity 50% to 70%), 4D-CT, or less commonly MRI, imaging can also help the surgeon preoperatively in those cases of reoperation.246,250
Surgical removal of the overactive parathyroid glands is the definitive treatment of PHPT in MEN1 patients, but the surgical approach remains controversial.251 Subtotal (3.5 gland) parathyroidectomy with thymectomy or total (four gland) parathyroidectomy with thymectomy and parathyroid autotransplantation to the forearm are the primary options for surgical therapy. Resection of less than three glands results in the highest risk of persistent and recurrent disease (OR 3.11, 95% CI = 2.00 to 4.84).252,253 A meta-analysis found that there was no significantly higher risk for persistent or recurrent HPT with a subtotal versus total procedure, but there was a significantly lower risk for permanent hypoparathyroidism with subtotal parathyroidectomy.252,254 In studies that followed MEN1 patients beyond 10 years, the recurrence rates for either procedure range from 40% to 60%255; recurrent hypercalcemia can be as high as 50% in autotransplanted tissue.216 Concomitant transcervical thymectomy is recommended to remove any potential ectopic parathyroid glands and to prophylactically remove the bulk of the thymus, which is at risk for the development of a thymic carcinoid.252,253,255,256
Surgical indications for treatment of asymptomatic PHPT in children with MEN1 are not clear,251


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


