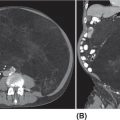
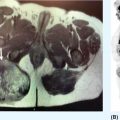
33726 Perivascular Epithelioid Cell Sarcoma Perivascular epithelioid cell neoplasms (PEComas) are a rare family of mesenchymal tumors, composed of histologically and immunohistochemically distinctive cells known as perivascular epithelioid cells (PECs) as defined by the World Health Organization. Clinically, many PEComas are benign and indolent in nature, but subsets of these tumors have a malignant potential and an aggressive course, and therefore it is important that these patients are identified and treated. PEComas have been described in case reports and series in a variety of different anatomic locations including the colon, pancreas, retroperitoneum, heart, adrenal gland, breast, eye, biliary tract, bone, urinary bladder, genitourinary tract, skull base, liver, uterus, cervix, skin, and soft tissues. This chapter reviews the epidemiology, clinical presentation, diagnostic approach, molecular drivers, and treatment options for PEC sarcomas. chemotherapy, clinical presentation, diagnostic approach, epidemiology, molecular drivers, perivascular epithelioid cell neoplasms, perivascular epithelioid cells, radiation therapy, treatment options, World Health Organization Diagnostic Techniques and Procedures, Drug Therapy, Epidemiology, Epithelioid Cells, Perivascular Epithelioid Cell Neoplasms, Radiotherapy, World Health Organization INTRODUCTION Perivascular epithelioid cell neoplasms (PEComas) are a rare family of mesenchymal tumors, composed of histologically and immunohistochemically distinctive cells known as perivascular epithelioid cells (PECs) as defined by World Health Organization (WHO).1 PEComas are notable for tumor cells that demonstrate a focal association with blood vessel walls and commonly express myogenic and melanocytic markers, including actin and/or desmin and HMB-45 and/or melan-A, respectively.1 This family of tumors consists of angiomyolipoma (AML), lymphangioleiomyomatosis (LAM), clear-cell “sugar” tumor of the lung and extra pulmonary sites (CCST), clear-cell myomelanocytic tumor of the falciform ligament/ligamentum teres, and rare histologically and immunophenotypically similar tumors arising from a variety of soft tissue and visceral sites.1–5 Pea and Bonetti et al. are credited with coining the term “perivascular epithelioid cell” in 1992 after first describing the presence of this unique cell with “prominent cytoplasmic borders and clear to granular, eosinophilic cytoplasm” in a perivascular distribution in both AML and CCST. They then suggested this cell was further distinguished in part by strong positivity of the melanocytic marker HMB-45, a common link among these rare tumors.2,5,6 Furthermore, genetic alterations in the tuberous sclerosis complex (TSC), leading to losses of TSC1 (9q34) or TSC2 (16p13.3) genes have been described in both sporadic cases and in patients with the autosomal dominant disease, tuberous sclerosis, which is characterized by mental retardation, seizures, and cellular proliferations, that is, AML and LAM.1,3,4 Clinically, many PEComas are benign and indolent in nature, but subsets of these tumors have a malignant potential and an aggressive course, and therefore it is important that these patients are identified and treated. Historically, treatment was mainly based on surgical resection as conventional doxorubicin-based chemotherapy typically employed in other soft tissue sarcomas (STS) has largely been ineffective. Given the rarity of PEComas, the majority of our understanding of the disease comes from small case series, retrospective analysis, and clinical experience. The identification of the alterations in TSC and its relationship to the mammalian target of rapamycin (mTOR) pathway has opened doors to the exploration of mTOR inhibitors as a means to treat these patients. In this chapter, we will review the epidemiology, clinical presentation, diagnostic approach, molecular drivers, and treatment options for PEC sarcomas. EPIDEMIOLOGY PEComas are exceedingly rare; in fact there is no Surveillance, Epidemiology, and End Results (SEER) data to track the benign or malignant PEComas.7 Per WHO publication as of 2013, there were approximately 200 reported cases of non-AML and non-LAM PEComas.1 Most commonly, PEComas are present in young to middle-aged adults (mean age 45 years). PEComas are markedly more frequent in females compared to males, with a female-to-male ratio of approximately 6:1. No links to a particular race or ethnicity have been reported.1,3,8 CLINICAL PRESENTATION AND NATURAL HISTORY PEComas are exceedingly rare and can present in a number of different soft tissue and visceral organs, leading to an array of presenting symptoms from asymptomatic to pain and bleeding, based on the primary location of the tumor. AML presenting classically of the kidney can occur in association with 338tuberous sclerosis or sporadically. AML cases associated with tuberous sclerosis are reported in both sexes with a female predominance, occurring in the third and fourth decades of life, as bilateral, small, multiple lesions, generally asymptomatic and benign. Sporadic cases tend to occur later in life, present as single lesions, unilateral, and larger. Complications can include renal failure and hemorrhage with a relatively low malignant potential.4 LAM, another member of this family of tumors, can occur sporadically, but is frequently seen in patients with TSC. This is a rare progressive disease affecting the lungs, predominately afflicting women. The natural history of the disease is a slow progression to respiratory failure with median survival of 8 to 10 years, and the only therapy is lung transplantation. CCST is generally a benign condition in which the vast majority of cases are sporadic.4,6 The remainder of this chapter will focus on the tumors in this family, which have come to be referred to in literature as PEComa not otherwise specified (PEComa-NOS). PEComas have been described in case reports and series in a variety of different anatomic locations including the colon,9–11 pancreas,12–14 retroperitoneum,15,16 heart,17 adrenal gland,18,19 breast,20 eye,21,22 biliary tract,23 bone,24,25 urinary bladder,26–28 genitourinary tract,29,30 skull base,31 liver,32–34 uterus,35,36 cervix,37,38 skin,39,40 and soft tissues.41 The clinical presentation varies based on anatomic location of the primary tumor. Patients presenting abdominopelvic PEComas describe progressive abdominal pain and weight loss. Patients presenting with genitourinary tract lesions frequently mention pain and hematuria as their chief complaint.29 Uterine and cervical tumors often present with vaginal bleeding and those of the gastrointestinal (GI) tract with GI bleeding. Our understanding of the natural history and prognostic features of these tumors is largely based on reviews of reported case studies and series. In 2005, Folpe et al. published the largest review to date at the time of 26 patients with PEComas of soft tissue and gynecologic origin.42 The median age was reported as 46 years (range, 15–97); there was a marked female predominance (22 females, four males), and the sites of involvement included omentum/mesentery (six cases), uterus (four cases), pelvic soft tissue (three cases), abdominal wall (two cases), uterine cervix (two cases), and vagina, retroperitoneum, thigh, falciform ligament, scalp, broad ligament, forearm, shoulder, and neck (one case each). They evaluated the characteristics of the tumors including size, histology (epithelioid, spindled, or mixed), grade, mitotic activity, presence of necrosis, and immunohistochemistry (IHC) results to assess the predictive and clinical features of these tumors. Of note, no patients in the series had a history of tuberous sclerosis. Based on their analysis, they suggested that PEComas could be classified as “benign,” “of uncertain malignant potential,” or “malignant.” High-risk features included size >5 cm, infiltrative growth pattern, high nuclear grade and cellularity, mitotic rate >1/50 high power fields (HPF), necrosis, and vascular invasion. Those classified as “benign” had <2 high-risk features and size <5 cm. Those classified in the “uncertain malignant potential” category had size ≥5 cm with no other high-risk features or nuclear pleomorphism/multinucleated giant cells only. Finally, those classified in the “malignant” category had two or more high-risk features. Recurrence and/or metastasis was strongly associated with tumor size >5 cm, mitotic activity greater than 1/50 HPF, and necrosis.42 In 2012, Bleeker et al. published a review of 234 cases of PEComa-NOS in English literature, evaluating information regarding diagnostic features, treatment approaches, and outcomes.2 Based on their multivariate analysis, primary tumor size ≥5 cm (p = .02) and high (1/50 HPF) mitotic rate (p < 0001) were the only factors significantly associated with recurrence following surgical resection.2 DIAGNOSTIC APPROCH High clinical suspicion is necessary for accurate diagnosis of these rare tumors. Like any soft tissue mass, early evaluation with imaging is critical to identify the primary location of the mass and metastatic disease. Evaluation with gadolinium-enhanced MRI and CT has been utilized in the diagnosis of these tumors. Referral to a multidisciplinary sarcoma center for initial management, including biopsy, pathology review, and surgical resection for localized disease is paramount for the best outcomes. Initial staging should include a high quality contrast-enhanced CT scan of the lungs, as well as abdomen, pelvis, and bone scan. PET/CT scan may be useful to identify occult lesions, particularly in advanced disease, but early lung metastases are often sub-centimeter in size and below the threshold of PET detection. RADIOGRAPHIC FEATURES On CT imaging, PEComas may appear heterogeneous with heterogeneous enhancement. On MRI imaging, they often exhibit low T1 signal and high T2 signal intensity with increased vascularity and 339heterogeneous enhancement.7 One review of 22 patients with hepatic PEComas described the lesions as well-defined, heterogeneous, arterial enhanced masses with dysmorphic vessels on CT, MRI, and ultrasound images.32 HISTOPATHOLOGIC FEATURES The hallmark of PEComas is the presence of the distinctive PECs. They are characterized by their perivascular location, often forming a radial arrangement of cells around the vascular lumen. PEComa-NOS tumors tend to comprise both epithelioid and spindle cell components in various degrees of proportion. The epithelioid cells are more commonly located in the immediate perivascular area, whereas the spindle cells resembling smooth muscle cells are often located away from the vessels. The PECs are described as having a clear to granular, lightly eosinophilic cytoplasm; small, centrally located, normochromatic, round to oval nuclei with small nucleoli; and slight atypia with sparse mitotic activity. However, a significant number of reported PEComas have increased mitotic activity and considerable atypical features, including nuclear atypia with hyperchromasia and irregularity.43 Immunohistochemically, PEC are characterized for expressing myogenic and melanocytic markers. The myogenic markers can include desmin, smooth muscle actin (SMA), muscle-specific actin/all muscle actin/HHF-35, muscle myosin and calponin. Desmin expression can be seen in approximately 30% of PEComas.8 The melanocytic markers include HMB-45, melan-A/ART-1, tyrosinase, and micropthalmia transcription factor. The most sensitive melanocytic markers for PEComa are HMB-45 and Melan-A. S-100 protein is reported as positive in approximately 30% of PEComas, and its presence does not necessarily imply a melanocytic malignancy.8 Cytokeratin expression is usually negative.43 Estrogen and progesterone receptors are frequently positive in renal AML, but have been rarely shown to be positive in PEComa-NOS.1,8,43 MOLECULAR AND BIOLOGIC DRIVERS Alterations in the TSC genes have been reported in both sporadic and in PEComas related to tuberous sclerosis. The most common abnormality observed is the loss of heterozygosity of the TSC2 gene on 16p13. Genetic alterations on TSC1 gene on 9q34 have also been appreciated. The TSC genes play an important role in regulation of the Rheb/mTOR/p70S6K pathway and therefore have been a focus for targeted therapy. Increasing studies suggest this pathway is complex with multiple protein components, but at the core of the pathway are four key proteins, including TSC1, TSC2, Rheb, and mTOR (mammalian target of rapamycin). Rheb protein is a member of the ras family of GTPases and when bound to guanosine triphosphate (GTP), it interacts with mTOR forming the activated complex termed mTORC1. This activation drives the mTOR/p70S6K pathway leading to downstream events, such as ribosome biogenesis, protein synthesis, and cell growth. Therefore, the loss of TSC1/TSC2 can have significant effects on the mTORC1 complex and result in cell growth. There is limited data on other genetic alterations observed in PEComas. TFE3 gene rearrangements have been increasingly observed in cases.20,44 Interestingly, 80% of TFE3 fusion negative PEComas have been shown to have TSC2 mutations, whereas those harboring TFE3 fusions lack TSC2 mutations.44,45 TREATMENT CONSIDERATIONS Complete surgical resection when feasible has historically been the mainstay of treatment. Surgery is not attempted in patients who are metastatic at the time of presentation or if the primary lesion is deemed unresectable. In 2012, Bleeker et al. reviewed 234 cases of PEComa, of which the majority, 95%, underwent surgical resection. Only 18% of patients had additional therapy beyond surgery. A minority of patients received neoadjuvant chemotherapy, radiotherapy, or combined chemoradiation with mixed response. Only one reported case had a robust response, the remaining either progressed or had minimal evidence of efficacy in the resected tumor. The role of adjuvant therapy is also unclear. The chemotherapy regimens typically utilized an anthracycline backbone typically used in other STS.2,43 In the metastatic setting, chemotherapy has shown little benefit. There has been enthusiasm for targeted therapies, most notably with mTOR inhibitors. Nanoparticle albumin-bound sirolimus (nab-sirolimus), an mTOR inhibitor, received Breakthrough Therapy designation from the Food and Drug Administration (FDA) based on preliminary data from the AMPECT trial presented at the American 340Society of Clinical Oncology (ASCO) 2019 meeting. This is the first prospective clinical trial in advanced PEComa to date. The trial was conducted in nine U.S. cities and enrolled 34 adult patients, 31 evaluable patients with confirmed PEComa. Involved sites included uterus, pelvis, retroperitoneum, lung, kidney, liver, brain, muscle, ovary, aorta, and small bowel. The primary objective was overall response rate (ORR). Interim analysis showed 42% had an interim analysis, 69% of the partial responses (PR) were ongoing, and the median progression-free survival (mPFS) was 8.9 months. Of the 25 patients with known mutational status, five had mutations in TSC1 and nine had mutations in TSC2. PR was achieved in 100% of patients with a TSC2 mutation (9/9), 20% (1/5) of those with a TSC1 mutation, and 9% (1/11) of patients without a mutation in TSC1 or TSC246 (NCT02494570). Anti-PD-1 therapy may play a role in the future. In one case report, a 46-year-old man with a PEComa located in the right popliteal fossa was initially treated with sirolimus, but was unable to tolerate the side effects. A year after his diagnosis, he presented with metastatic disease to the lungs, rib cage, and a fungating mass in the right popliteal fossa. He underwent an amputation and video-assisted thorascopic surgery (VATs) to confirm metastatic disease. He was subsequently treated with pazopanib and progressed. He was subsequently started on nivolumab, a PD-L1 inhibitor and imaging done at 2 and 5 months revealed significant disease response. At 12 months, he was progression-free with no identifiable metastatic lesions.47 SUMMARY PEComas are an exceedingly rare subset of STS. They vary widely in clinical behavior from benign to aggressively malignant lesions. Significant progress has been made with regard to the molecular mechanism underlying the disease and understanding of histopathology and IHC for disease diagnosis. The risk assessment remains imprecise, but large tumors >5 cm and high mitotic rate of 1/50 HPF are widely accepted as risk factors for more aggressive disease. The mainstay of treatment remains surgical resection when feasible. In recurrent/metastatic disease, chemotherapy and radiation have been used with mixed to minimal benefit. The first prospective clinical trial has led to the first breakthrough therapy designation by the FDA for nab-sirolimus in the management of these patients. More needs to be learned about these unique tumors.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


